

Research Article
Preparation, Structural Characterization and Functional Properties of Chitosan Derivative
Institute of Agro-products Processing Science and Technology, Chinese Academy of Agricultural Sciences, PR China
*Corresponding author: Yi-Ming Ha, Institute of Agro-products Processing Science and Technology, Chinese Academy of Agricultural Sciences, PR China, Tel: +86010-62815971, Fax: +86010-62829727, E-mail: hayiming@sina.com
Received: March 28, 2017 Accepted: April 18, 2017 Published: January 5, 2018
Citation: Guo Q, Cui WH, Yang Y, Li QP, Ha YM. Preparation, Structural Characterization and Functional Properties of Chitosan Derivative. Madridge J Food Technol. 2018; 3(1): 84-90. doi: 10.18689/mjft-1000113
Copyright: © 2018 The Author(s). This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Chitosan-citric acid (CTS-CA), was prepared using chitosan (CTS) and citric acid (CA) with sodium hypophosphite (SHP) as a catalyst. The preparation conditions for the CTS-CA were as follows: mole ratio of CA and SHP was 10:1, reaction time was 3 h, reaction temperature was 110°C, and mass ratio of CA and CTS was 2.5:1. In such conditions, the overall yield and the weight-average molar mass of CTS-CA were 48.62% and 529,000, respectively, and its water solubility was higher than that of CTS. The structure of CTS-CA was characterized by FTIR, SEM, TGA, XRD and NMR. Results showed that the acylation reaction happened between CTS and CA, and CTS-CA was synthesized, and acylation mechanism was proposed. In addition, CTS-CA exhibited good effect on adsorping cadmium ions and scavenging free radicals.
Keywords: Chitosan; Acylation reaction; Chitosan-Citric acid; Structural characterization; Functional properties.
Introduction
Chitosan (CTS), a partially deacetylated derivative of chitin under alkaline conditions, is a linear polymer compound containing 2-amino-2-deoxidation-β-D-glucose linked by β-1,4-glycosidic bonds [1]. It has been widely used in food [2], medicine [3], environmental protection [4], textile [5] and cosmetic industries [6,7], due to non-toxic and biodegradable. CTS has ability to chelate metal ions and antibacterial activity [8,9]. However, CTS exhibits poor water solubility, and only dissolves in selective dilute acid solutions because of the intermolecular or intramolecular hydrogen bonds between amino and hydroxyl groups. This high crystallinity limites CTS application. Thus, different physical and chemical methods were developed to prepare various CTS derivatives to improve its water solubility with quality features [10-12].
Carboxymethylation, acylation, quaternary ammoniation and esterification methods has been used to modify CTS to improve its water solubility [13-15]. Chung et al. showed that the solubility of modified CTS derivatives by Maillard reaction was siginificantly greater than that of CTS [11]. In addition, O-PEGylated chitosan was soluble in water and aqueous solutions of wide pH range [16]. But reaction conditions of carboxymethylation was difficult to control and chloroactic acid used in reaction process was highly toxic substance [17]. Quaternary ammoniation and esterification produced a lot of acid or alkali waste liquid, resulting in the pollution of environment [17,18]. Some modified methods were complex and needed long preparation time [11]. Therefore, it was necessary to develop a relatively simple and safe method for improving the water solubility of CTS.
Citric acid (CA) has a good hydrophilicity and its carboxyl has high reactivity with amino group [19]. In the national standards, it is a kind of food additive and its usage is safe within the prescribed scope and dosage [20]. Moreover, it can be decomposed into water and carbon dioxide (non-toxicity) heating at temperature above 175°C [21]. However, the preparation of CTS derivative using CTS and CA has not been reported. The aim of this study was to synthesize CTS-CA through acylation reaction of CA and CTS with sodium hypophosphite (SHP) as a catalyst. The structural of CTS-CA was characterized by FTIR, SEM, TGA, XRD and NMR. The water solubility, and adsorption and scavenging properties of CTS-CA were investigated.
Materials and Methods
Materials
CTS (food grade) was purchased from Shanghai Seebio Biotech, Inc. The weight-average molar mass of CTS was 261,000, as determined using a laser scattering instrument. The deacetylation degree of CTS was 84.40%, as determined using linear potentiometric tiration method. CA, acetic acid (guaranteed reagent), SHP, salicylic acid and iron (II) sulfate heptahydrate (FeSO4·7H2O) were purchased from China National Pharmaceutical Group Corporation. Sodium acetate anhydrous (guaranteed reagent) was purchased from Aladdin. Dextran (guaranteed reagent) and DPPH were purchased from Sigma. Absolute ethanol was purchased from Beijing Chemical Works. Cadmium piece, nitric acid (BV-III), and hydrogen peroxide (BV-III) were purchased from Beijing institute of chemical reagents. All reagents were analytically pure unless otherwise specified.
Preparation of CTS-CA
CTS-CA was prepared via the acylation reaction method by mixing 1.50 g of CTS with 30 g of water containing different amounts of CA (from CA 4 g/g to 1 g/g of CTS) and SHP as the catalyst (from CA 10 mol/mol to 1 mol/mol of SHP). The reaction mixture was transferred to a pressurized bottle, and placed in a circulating air oven at different reaction temperatures (70-150°C) for different reaction times (2-4 h). Samples were precipitated and purified by washing with 85% ethyl alcohol after cooling to room temperature, suction filtration, and then vacuum freeze drying at -50°C for 36 h. The purified products were stored prior to analysis.
Yield of products
The overall yield of product was calculated using Eq. (1)
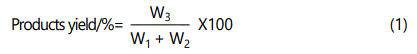
W1: mass of CTS (g); W2: mass of CA (g); W3: mass of CTS-CA (g).
Weight-average molar mass measurement
The weight-average molar mass of CTS-CA was determined by multi-angle laser light scattering instrument with size exclusion chromatography (MALLS-SEC), including a multi-angle laser detector (DAWN HELEOSII, Wyatt, USA), a UV detector (L-2400, HITACHI, Japan), and a differential refractometer (WREX-14, Wyatt, USA). Dextran (from Leuconostoc spp., Sigma) was used as standard, with a relative molecular mass (Mr) of 40,000.
The prepared CTS-CA (1 mg/mL) was dissolved in a moving phase (0.1 mol/L acetic acid and sodium acetate buffer solution (pH 4.5) containing 0.2‰ NaN3) and filtered using a 0.45 µm membrane. The injection volume was 200 µL, the UV detector wavelength was 280 nm, the differential refractometer detector wavelength was 690 nm and the flow rate was 0.50 mL/min.
Water solubility measurement
About 0.10 g of CTS and CTS-CA were dissolved separately in 10 mL of distilled water and mixed well. Water solubility was observed with the naked eyes.
Infrared spectrometry
A Brucker Tensor37 FTIR spectrometer (BRUKER OPTICS, Germany) was used to measure infrared spectra. The sample was mixed with potassium bromide (KBr) (about 200-400 mg), placed in a sampling cup, flattened and compressed into a transparent flake using a tablet machine at 8 t pressures. The powdered sample was placed in the light path at room temperature with KBr pellet as reference to determine the spectrum in the range of 4000-400 cm-1.
SEM determination
Samples were subjected to a gluing machine and metal spraying, their surface morphologies were studied through SEM (FEI, America, Quanta 200 FEG) under high vacuum at a voltage of 6 kV. Different magnified photographs were obtained.
Determination of TGA
TGA was performed with the Perkin-Elmer TG/DTA analyser (Pyris-115, USA). Samples were weighed in an aluminium alloy crucible and then placed in the analyser. An empty aluminium crucible was used for reference. TGA experimental conditions were nitrogen atmosphere of 50 mL/min, the heating rate of 10°C/min, sample weight of 1.5-2.0 mg and scanning temperatures range of 40-550°C.
Powder XRD measurement
The XRD patterns of samples were obtained using an X-ray diffractometer (XD-2 XRD, Rigaku Dmax/2400 Neo-Confucianism Instrument, Japan) with Cu Kα radiation (λ=1.50 Å at 40 kV, 100 mA in the 2 theta range of 5° to 60°, scan speed of 6°/min and use of graphite monochromator).
NMR spectroscopy
Samples were characterised using 13C spectroscopy on a Bruker 400 MHz WB solid-state NMR spectrometer (AVANCE III) at a resonance frequency of 100 MHz. All samples were ground and crushed before usage. Measurements were performed at room temperature.
Cadmium ion adsorption experiment
0.10 g CTS-CA sample was mixed in 50 mL Cd2+ solution (25 mg/L), then adjusted pH of the solution to 2-10, shocked 8 h, centrifuged for 10 min (4,000 r/min) at 4°C. 1 mL of supernatant liquid, 6 mL of HNO3 and 2 mL of H2O2 were mixed, digested, cooled and capacity to 100 g, then determined by Inductively Coupled Plasma Mass Spectrometry (ICP-MS). Measurement conditions were as follows: high frequency incident power was 1550 W, atomizing chamber temperature was 2°C, carrier gas flow rate was 1.07 L/min, peristaltic pump was 0.10 rps, lens voltage was 10.20 V, integration time was 0.10 s, sampling period was 0.31 s and scanning number was 3. Adsorption capacity and adsorption rate were calculated using Eq. (2) and (3)


Q: Adsorption capacity (mg/g); η: Adsorption rate (%); V: Volume of solution (mL); C0: Cd2+ concentration before adsorption (mg/L); C: Cd2+ concentration after adsorption (mg/L); W: Mass of CTS-CA (g).
Removing experiment of hydroxyl free radical
The hydroxyl free radical clearance rate was measured according to the method of Liu et al. [22]. with some modifications. 1 mL of CTS-CA solution (0.2-1.6 mg/mL), 1 mL of FeSO4 (9 mmol), 0.50 mL of salicylic acid-ethanol solution (9 mmol) and H2O2 (0.15%) were mixed, heated at 37°C water bath for 30 min, and then measured at 510 nm by ultraviolet spectrophotometer. The hydroxyl free radical clearance rate was calculated using Eq. (4)
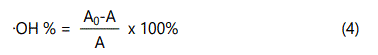
A0: Absorbance of control group; A: Absorbance of sample groups.
Removing experiment of DPPH free radical
The DPPH free radical clearance rate was determined by a modified method of Shimada et al. [23]. 2 mL DPPH-ethonal solution (0.1 mmol) and 1.8 mg/mL CTS-CA solution (1-6 mL) were mixed, heated at 25°C water bath for 30 min, and then measured at 517 nm after cooling by ultraviolet spectrophotometer. The DPPH free radical clearance rate was calculated using Eq. (5)
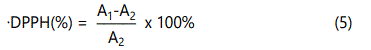
A1: Absorbance of control group; A2: Absorbance of sample groups.
Results and Discussion
CTS-CA preparation conditions analysis
The effects of mole ratio of CA and SHP, reaction time, reaction temperature, and mass ratio of CA and CTS on the yield and weight-average molar mass of CTS-CA were shown in figure 1. When the experiment conditions were reaction time of 3 h, reaction temperature of 110°C, and CA and CTS mass ratio of 2:1, the yield of CTS-CA initially increased and then decreased gradually (Figure 1a). It may be attributed to the reduction of CA compared to that of SHP, which made the balance moving in the direction of reverse reaction. The weight-average molar mass of CTS-CA increased rapidly, peaked at CA and SHP mole ratio of 10:1, and then remained unchanged with decreasing of CA and SHP mole ratio. The reason for explaining was that CTS-CA was accumulated after CTS reacting with CA in the presence of SHP, and then achieved saturated state as the reaction proceeding. These results indicated that the yield and weight-average molar mass of CTS-CA attained the highest at CA and SHP mole ratio of 10:1.
As shown in figure 1b, the preparation conditions for the CTS-CA were CA and SHP mole ratio of 10:1, reaction temperature of 110°C, and CA and CTS mass ratio of 2:1. The yield of CTS-CA increased with the increase of reaction time, remained unchanged at 2.5 h-3 h, and then raised rapidly. The weight-average molar mass increased with the increase of reaction time, then decreased with the extension of reaction time. The reason for this phenomenon may be due to degradation of CTS-CA with the extension of reaction time. The yield and weight-average molar mass of CTS-CA were better at reaction time of 3 h.
When the preparation conditions were CA and SHP mole ratio of 10:1, reaction time of 3 h, and CA and CTS mass ratio of 2:1 (Figure 1c), no obvious changes were observed on the yield of CTS-CA at reaction temperature lower than 100°C. However, it decreased significantly at the temperature higher than 100°C, indicating that this reaction was sensitive to high temperature (≥100°C), and reaction temperature should be strictly controlled. Under the same condition, the weightaverage molar mass of CTS-CA gradually increased with the heating temperature, peaked at 110°C, and then decreased. Thus, the reaction temperature for preparing CTS-CA was chose at 110°C.
At optimizational CA and SHP mole ratio of 10:1, reaction time of 3 h, and reaction temperature of 110°C, the yield of CTS-CA decreased with the increasing concentration of CA (Figure 1d). The weight-average molar mass of CTS-CA increased slightly, reached maximum at CA and CTS mass ratio of 2.5:1, and then decreased.
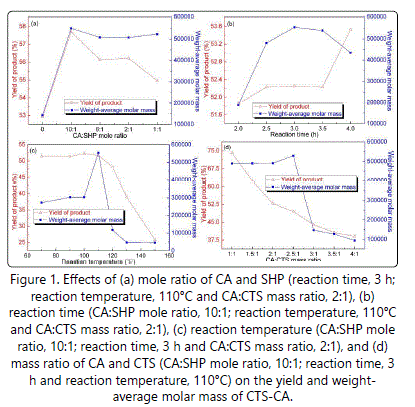
Based on the above results, the optimal preparation conditions for CTS-CA were as follows: CA and SHP mole ratio was 10:1, reaction time was 3 h, reaction temperature was 110°C and mass ratio of CA and CTS was 2.5:1. And in these conditions, the overall yield and weight-average molar mass of CTS-CA were 48.62% and 529,000, respectively.
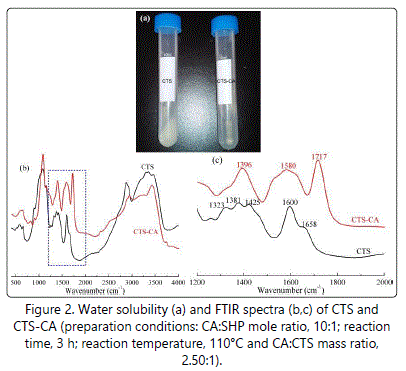
Water solubility analysis
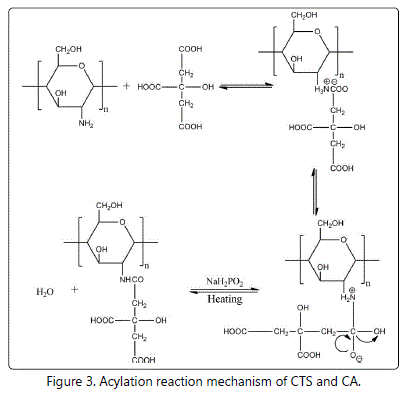
As shown in figure 2a, the water solubility of CTS-CA was better than that of CTS, indicating that the chemical reaction between CTS and CA occured. This reaction not only destroyed intermolecular and intermolecular hydrogen bonds of CTS, but also introduced the hydrophilic group (carboxyl). The speculated reaction mechanism of CTS and CA was shown in figure 3. Firstly, CTS and CA formed a proton salt after mixturing, the transition state was generated by intermolecular rearrangement, and then the water molecule was taken off under the condition of the catalyst and heating, leading to the formation of CTS-CA [24-26].
FTIR analysis
The FTIR spectra of CTS and CTS-CA were shown in figure 2b and 2c. Raw CTS showed signals at 1323 cm-1, 1381 cm-1, 1425 cm-1, 1600 cm-1 and 1658 cm-1 (Figure 2c), which were attributed to symmetrical angular deformation of C-N stretching, N-H plane bending deformation, C-N stretching coupled with N-H bending of amino groups and NH2 angular vibration, respectively [1,6]. Compared with the FTIR spectrum of CTS, the spectrum of CTS-CA showed a new absorption peak at around 1717 cm-1, which corresponded to C=O of CA, while the absorption at 1473-1673 cm-1 appeared as a wide absorption peak including N-H plane bending vibration of secondary amide at 1535-1560 cm-1 and C=O stretching vibration of secondary amide at 1635-1650 cm-1 (Figure 2c). The absorption peak for C-OH plane bending of CA appeared at 1396 cm-1 (Figure 2c). The FTIR spectra indicated that acylation reaction occured between the amino group of CTS and the carboxyl group of CA, confiring the mechanism of CTS and CA shown in Figure 3.
SEM analysis
The structural morphologies of CTS and CTS-CA were differences (Figure 4). The SEM of CTS showed large particle sizes and a smooth and compact structure (Figures 4a-4c), while CTS-CA displayed smaller particle sizes and a porous structure (Figures 4d-4f). Such differences in the structure of CTS and CTS-CA could be attributed to chemical modification. It explained the higher solubility of CTS-CA than that of CTS, due to porous structure that was advantageous to the water molecules to enter.

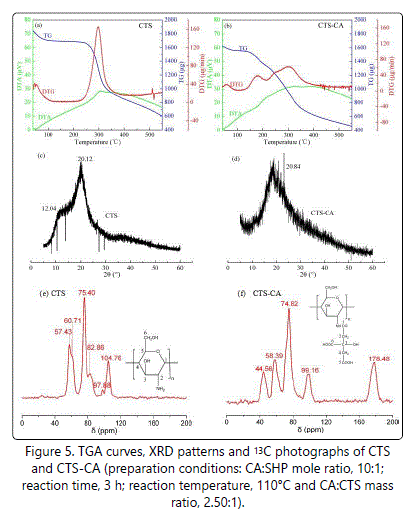
Thermogravimetric analysis
Thermogravimetric analysis (TGA) was carried out to determine the thermal properties and stability of the new material. The DTA curves of CTS (Figure 5a) and CTS-CA (Figure 5b) showed that their decomposition reaction were endothermic process. The TG curves showed that the thermal stability of CTS-CA was lower than that of CTS (Figures 5a and 5b). The initial degradation temperature of the CTS and CTS-CA were 239.5°C (Figure 5a), and 129.8°C (Figure 5b). The lower stability of CTS-CA may be attributed to the less intermolecular and intramolecular hydrogen bonds compared to that of CTS. Based on the DTG curve of CTS (Figure 5a), the maximum weight loss for CTS was 165 µg/min at 297.10°C. The DTG curve of CTS-CA showed that the weight loss of CTS-CA was divided into three stages, 39.10 µg/min at 177.60°C, 48.10 µg/min at 250.10°C, and 61.60 µg/min at 297.40°C, respectively. The peak of weight loss at 177.60°C appeared to the carboxyl decomposition of CA because boiling point of CA was 175°C (Figure 5b). The TGA results also confirmed that CTS-CA was synthesized successfully.
Powder XRD analysis
The powder X-ray diffraction patterns of CTS and CTS-CA were given in figure 5c and 5d, respectively. The CTS (Figure 5c) exhibited two typical peaks at 2θ=12.04° and 20.12°. Huang et al. [10]. reported that the reflection at 2θ=12.04° was assigned to the crystal form I and attributed to the hydrated crystals with low crystallinity. The strongest intensity at 2θ=20.12° represented the crystallinity of the crystal form II. For CTS-CA (Figure 5d), a relatively obtuse and weak broad peak appeared at 20.84°, indicating that the ability of CTS to form hydrogen bonds may reduce after chemical modification of the amino groups at the C2 position: as such, a smaller fraction of crystalline phase and a larger fraction of amorphous phase were formed [12,27-29]. It was suggested that the crystallinity of CTS-CA decreased, and this decrease in crystallinity of CTS-CA was attributed to the deformation of the strong hydrogen bond in the free chitosan molecular and also due to the substitution of CA on it [4,14,15]. It better explained CTS-CA dissolving in water more easily than that of CTS due to their crystallinity, because the higher the crystallinity of substance, the more difficult it is to dissolve generally.
Solid-state C NMR analysis
The high resolution solid state 13C NMR was used to characterisation of new substances. The 13C NMR spectra of CTS and CTS-CA obtained in this study were shown in Figures 5e and 5f. In the spectrum of CTS (Figure 5e), the peak at 104.76 ppm, 97.88 ppm, 82.86 ppm, 75.40 ppm, 60.71 ppm and 57.43 ppm were attributed to the C1, C2, C3, C4, C5, C6 of CTS, respectively [30]. Compared with CTS, the 13C NMR spectrum of CTS-CA showed additional peaks at 178.48 ppm (Figure 5f C7 or C8), which was attributed to the carbonyl carbon atoms of CA present in the CTS-CA. However, chemical shift of CTS-CA (Figure 5f C2) moved to low field direction at 99.16 ppm compared with CTS (Figure 5e C2 97.88 ppm), it may be due to high electronegativity of carbonyl oxygen existed in amide bond transformed amino group of C2. The differences in the nature and intensity of peaks within the region of 40-100 ppm of CTS-CA from those of CTS indicated that the additional aliphatic (CA) carbon atoms were present in the CTS-CA [4,31,32]. These evidences obviously supported that the amino groups of the raw chitosan were converted to amides.
Compared with the CTS, the CTS-CA exhibited high water solubility. In addition, the FTIR, TGA and solid state 13C NMR analyses indicated that the hydrophilic group (carboxyl) was successfully introduced into CTS via acylation reaction. Meanwhile, XRD and SEM figures showed that its crystal construction was destroyed, presenting porous structure, which was more advantageous to water molecules to enter. The results showed that the structures of CTS-CA were considerably different from CTS.
Cadmium ion adsorption analysis
The adsorption of Cd2+ was significantly different under the condition of different pH (Figures 6a and 6b). Concentration of Cd2+ in the solution gradually decreased, and then remained basically unchanged with the increase of pH. In other words, with the increase of pH, adsorption capacity and adsorption rate gradually increased, and then maintained essentially constant. The adsorption capacity and adsorption rate reached maximum at pH of 9, 1241.95 mg/g and 98.21%, respectively (Figure 6b), which were much better than that of Yan et al. (87.74 mg/g and 81.22%) [33]. The possible reason was that side chain carboxyl group of CTS-CA forming COO-in alkaline conditions was easier to combine with Cd2+.
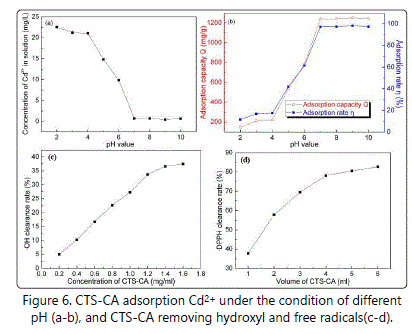
Hydroxyl free radical and DPPH free radical clearance analysis
According to figure 6c, CTS-CA had a certain ability to remove hydroxyl free radical and its eliminated capability gradually enhanced with the increase of CTS-CA concentration. DPPH clearance rate increased with the increase of the volume of CTS-CA (1.8 mg/mL), suggesting that CTS-CA had a good ability to eliminate DPPH free radical (Figure 6d). Therefore, CTS-CA may be has a certain antioxidant effect.
The structures and properties are closely linked, so structural changes inevitably lead to changes in property. Compared with CTS, CTS-CA had high water solubility, and exhibited better effect on adsorption of cadmium ion and scavenging free radicals (·OH and ·DPPH), which may become a valuable material.
Acknowledgements
This work was financially supported by the “National Science and Technology Supported Program for the Control of Food Biological Toxin Pollution and the Early Warning Technology of the Radioactive Pollution” (Grant No. 2012BAK17B13) and the “Special Fund for Agro-scientific Research in the Public Interest” (Grant No. 201103007).
References
- Duan WG, Shen CM, Fang HX, Li GH. Synthesis of dehydroabietic acidmodified chitosan and its drug release behavior. Carbohyd Res. 2009; 344(1): 9-13. doi: 10.1016/j.carres.2008.08.007
- Aider M. Chitosan application for active bio-based films production and potential in the food industry: Review. Lebensm Wiss Technol. 2010; 43(6): 837-42. doi: 10.1016/j.lwt.2010.01.021
- Chakraborty SP, Sahu SK, Pramanik P, Roy S. Biocompatibility of folatemodified chitosan nanoparticles. Asia Pac J Trop Biomed. 2012; 2(3): 215-19. doi: 10.1016/S2221-1691(12)60044-6
- Krishnapriya KR, Kandaswamy M. A new chitosan biopolymer derivative as metal-complexing agent: synthesis, characterization, and metal (II) ion adsorption studies. Carbohyd Res. 2010; 345(14): 2013-022. doi: 10.1016/j.carres.2010.06.005
- Vellingir K, Ramachandran T, Senthilkumar M. Eco-Friendly application of nano-chitosan in antimicrobial coatings in the textile industry. Nanosci Nanotech Lett. 2013; 5(5): 519-29. doi: 10.1166/nnl.2013.1575
- Yenilmez E, Başaran E, Yazan Y. Release characteristics of vitamin E incorporated chitosan microspheres and in vitro–in vivo evaluation for topical application. Carbohyd Polym. 2011; 84(2): 807-11. doi: 10.1016/j.carbpol.2010.07.002
- Xie WM, Xu PX, Wang W, Liu Q. Preparation and antibacterial activity of a water-soluble chitosan derivative. Carbohyd Polym. 2002; 50(1): 35-40. doi: 10.1016/S0144-8617(01)00370-8
- Aytekin AO, Morimura S, Kida K. Synthesis of chitosan-caffeic acid derivatives and evaluation of their antioxidant activities. J Biosci Bioeng. 2011; 111(2): 212-16. doi: 10.1016/j.jbiosc.2010.09.018
- Huang XY, Mao XY, Bu HT, Yu XY, Jiang GB, Zeng MH. Chemical modification of chitosan by tetraethylenepentamine and adsorption study for anionic dye removal. Carbohyd Res. 2011; 346: 1232-1240.
- Chung YC, Tsai CF, Li CF. Preparation and characterization of watersoluble chitosan produced by Maillard reaction. Fisheries Sci. 2006; 72(5): 1096-1103. doi: 10.1111/j.1444-2906.2006.01261.x
- Kaczmarek H, Zawadzk J. Chitosan pyrolysis and adsorption properties of chitosan and its carbonizate. Carbohyd Res. 2010; 345(7): 941-47. doi: 10.1016/j.carres.2010.02.024
- Singh N, Kayastha AM. Cicer α-galactosidase immobilization onto chitosan and Amberlite MB-150: optimization, charactrization, and its applications. Carbohyd Res. 2012; 358: 61-66. doi: 10.1016/j.carres.2012.06.013
- Anisha GS, Prema P. Reduction of non-digestible oligosaccharides in horse gram and green gram flours using crude α-galactosidase from Streptomyces griseoloalbus. Food Chem. 2008; 106(3): 1175-179. doi: 10.1016/j.foodchem.2007.07.058
- Hamer SN, Moerschbacher BM, Kolkenbrock S. Enzymatic sequencing of partially acetylated chitosan oligomers. Carbohyd Res. 2014; 392: 16-20. doi: 10.1016/j.carres.2014.04.006
- Gorochovceva N, Makuška R. Synthesis and study of water-soluble chitosan-O-poly (ethylene glycol) graft copolymers. Eur Polym J. 2004; 40(4): 685-91. doi: 10.1016/j.eurpolymj.2003.12.005
- Xu Y, Du Y, Huang R, Gao L. Preparation and modification of N-(2-hydroxyl) propyl-3-trimethyl ammonium chitosan chloride nanoparticle as a protein carrier. Biomater. 2003; 24(27): 5015-022. doi: 10.1016/S0142-9612(03)00408-3
- GB 2760-2011. National food safety standards of using food additives. Ministry of health of the Peopleʼs Republic of China. 2011.
- Yang J, Webb AR, Ameer GA. Novel Citric Acid-based biodegradable elastomers for tissue engineering. Adv Mater. 2004; 16(6): 511-16. doi: 10.1002/adma.200306264
- Liu J, Jia L, Kan J, Jin CH. In vitro and in vivo antioxidant activity of ethanolic extract of white button mushroom (Agaricus bisporus). Food Chem Toxicol. 2013; 51: 310-16. doi: 10.1016/j.fct.2012.10.014
- Shimada K, Fujikawa K, Nakamura T. Antioxidative properties of xanthan on the autoxidation of soybean oil in cyclodextrin emulsion. J Agr Food Chem. 1992; 40(6): 945-48. doi: 10.1021/jf00018a005
- Yang CQ, Chen D, Guan J, He Q. Cross-linking cotton cellulose by the combination of maleic acid and sodium hypophosphite. 1. Fabric wrinkle resistance. Ind Eng Chem Res. 2010; 49(18): 8325-332. doi: 10.1021/ie1007294
- Ye T, Wang B, Liu J, Chen J, Yang Y. Quantitative analysis of citric acid/sodium hypophosphite modified cotton by HPLC and conductometric titration. Carbohyd Polym. 2015; 121: 92-98. doi: 10.1016/j.carbpol.2014.12.028
- Wang W, Yu W. Preparation and characterization of CS-g-PNIPAAm microgels and application in a water vapour-permeable fabric. Carbohyd Polym. 2015; 127: 11-18. doi: 10.1016/j.carbpol.2015.03.040
- Wang JP, Chen YZ, Zhang SJ, Yu HQ. A chitosan-based flocculant prepared with gamma-irradiation-induced grafting. Biores Technol. 2008; 99(9): 3397-402. doi: 10.1016/j.biortech.2007.08.014
- Jiang M, Wang K, Kennedy JF, Nie J, Yu Q, Ma G. Preparation and characterization of water-soluble chitosan derivative by Michael addition reaction. Int J Biol Macromol. 2010; 47: 696-699. doi: 10.1016/j.ijbiomac.2010.09.002
- Jablońska-Pikus T, Charmas W, Gawdzik B. Synthesis and characterization of methacrylate polymeric packings based on bisphenol-S. J Appl Polym Sci. 2000; 75(1): 142-48. doi: 10.1002/(SICI)1097-4628(20000103)75
- Loubaki E, Ourevitch M, Sicsic S. Chemical modification of chitosan by glycidyl trimethylammonium chloride. Characterization of modified chitosan by 13C- and 1H-NMR spectroscopy. Eur Polym J. 1991; 27(3): 311-17. doi: 10.1016/0014-3057(91)90111-Z
- Kono H, Teshirogi T. Cyclodextrin-grafted chitosan hydrogels for controlled drug delivery. Int J Biol Macromol. 2015; 72: 299-308. doi: 10.1016/j.ijbiomac.2014.08.030
