

Research Article
Physiochemical and Conformational Properties of Soluble Aggregates from Soy Protein Isolates Mediated by Hydrothermal Cooking: A comparative Study with Moisture Heat Treatment
1Department of Life Science and Food Technology, Hanshan Normal University, PR China
2Department of Food Science and Technology, South China University of Technology, PR China
3Haikou Experimental Station, Chinese Academy of Tropical Agricultural Science (CATAS), PR China
*Corresponding author: Xiao-quan Yang, Professor, Department of Food Science and Technology, South China University of Technology, PR China, Tel: +86-20-87114262, Fax: +86-20-87114263, E-mail: 10282361@qq.com
Received: April 20, 2016 Accepted: June 28, 2016 Published: October 3, 2016
Citation: Zhang B, Yuan DB, Yang XT, et al. Physiochemical and Conformational Properties of Soluble Aggregates from Soy Protein Isolates Mediated by Hydrothermal Cooking: A comparative Study with Moisture Heat Treatment. Madridge J Food Technol. 2016; 1(1): 1-9. doi: 10.18689/mjft-1000101
Copyright: © 2016 The Author(s). This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Two kinds of soluble aggregates, HC-SPI and HT-SPI, were obtained through centrifugation at 10000 g for 10 min, from two modified soy protein isolates, which were mediated by hydrothermal cooking at 121°C (0.1 MPa, gauge pressure) for 30 min in an autoclave and by moisture heat treatment at 90°C for 30 min in a water bath, respectively. The physicochemical and conformational properties, including SDS-PAGE/GPC, surface charge and hydrophobicity, free sulfhydryl group (SH) contents and disulfide bond (SS) contents, as well as secondary and tertiary conformations, were evaluated. Remarkable differences in SDS-PAGE patterns, GPC profiles, surface charge and hydrophobicity, free sulfhydryl group (SH) and disulfide bond (SS) contents, tertiary and quaternary conformations were observed between HC-SPI and HT-SPI. The significantly different free sulfhydryl group (SH) contents between HC-SPI and HT-SPI (especially as compared to N-SPI) suggest discrepancy in S-S formation mechanism. Both of the tertiary and quaternary conformations showed varying exposure extent with varying aromatic residues (Phe, Tyr and Trp).These results suggested good relationships between the physicochemical properties and conformational features of these soluble aggregates from SPI.
Keywords: Soy protein isolates; Modification; Soluble aggregates; Hydrothermal cooking; Heat treatment.
Introduction
Soy proteins have been widely used as an important food ingredient in a lot of food formulations, due to its high nutritional and functional properties. Soy protein isolate (SPI) is one of the most important commercially available soy protein products [1]. Glycinin (11S) and β-conglycinin (7S) globulins are the major components of soy protein isolates [2]. They must possess appropriate functional properties for food applications. These properties are influenced by the composition, structure, and conformation of ingredient proteins [3].
Soy protein isolates can be modified by different treatments to improve specific functional properties, heating being one of the more frequently used for that purpose [4]. Heat-induced aggregating of soy proteins gives them new properties such as solubility, emulsification, and foaming that can be useful in food products [3,5-7]. Gelling of proteins is induced by the unfolding and the subsequent aggregation of proteins [8]. When heated higher than the denaturing temperature, the hydrophobic groups buried in the native state are exposed to the surface, and as the surface hydrophobicity increases, net charge decreases. The changes of structure lead to the gel network formation [9]. Furthermore, soy aggregates had been identified to stabilize the emulsion because the adsorbed layers on oil droplet consisted of aggregated proteins [10]. It has been demonstrated that the functionality depends basically on the degree of dissociation, denaturation, and aggregation of the glycinin and β-conglycinin [11-13]. The subunits of glycinin and β-conglycinin could form some kind of soluble aggregates [11,14-16] and could also form some kind of insoluble aggregates [17] by moisture heat treatment. Discrepancy in the structure-function relationship of soluble and insoluble aggregates was also reported [17].
Hydrothermal cooking, a steam-infusion treatment often known as jet cooking, was employed to refunctionalize ethanol denatured soy protein concentrates, and gained significant increase in its major functional properties [18]. And the refunctionalization of the heat-denatured soy proteins could also be achieved through this method [19]. Some previous literatures also adopted autoclaves to serve as hydrothermal cooking for refunctionalization of proteins [20]. In our previous study using alcohol-washed insoluble soy protein concentrates (SPC) as the starting material, soluble aggregates with good functionality were formed from those insoluble soy protein components by hydrothermal cooking in an autoclave system [21].
To our knowledge, as for modified proteins by heating, such as moisture heat treatment or hydrothermal cooking, the induced aggregates may play key roles in conveying good functionality. To date, the aggregates mediated by heat treatment were extensively studied [4,17,22], but literatures concerning about the properties of the induced soluble aggregates by hydrothermal cooking, especially the conformational features, were scarcely reported. Thus, the main objective of this study was to compare the physicochemical and conformational properties of soluble aggregates, mediated by hydrothermal cooking and heat treatment, taking soy protein isolates as the starting materials.
Materials and Methods
Materials
Low denatured defatted soy flakes were provided by Shandong Yuwang Industrial & Commercial Co., Ltd. Soy flakes were obtained by dissolving through flash evaporation and toasted at about 60°C under vacuum to avoid heat denaturation. The flakes contained protein content 56.5% (N×6.25, dry basis) and nitrogen solubility index 85%. All chemical reagents used were of analytical or better grade.
Preparation of soybean protein isolates (SPI)
Defatted soybean seed meal was mixed with 10-fold (w/v) deionized water, and the mixture was adjusted to pH 8.5 with 1.0 mol/L NaOH. After 2hrs, the resulting suspension was centrifuged at 8000×g for 30 min to remove the insoluble material. Then, the pH of the supernatant was adjusted to pH 4.8 at 4°C with 2 mol/L HCl, and the precipitate or curd was collected by centrifugation (8000×g, 10 min). The protein precipitate was washed with pre-cooled deionized water and dispersed in deionized water, and dialyzed three times (each time for about 8hrs) at 4°C against 0.01 mol/L Tris-HCl buffer, pH 7.0 (1:100, v/v). The protein content of the SPI solution was determined by Kjeldahl method (N×6.25), and adjusted to 1.0 g/100 mL with the same buffer. The protein solution was directly freeze-dried to produce SPI samples.
Modification of SPI by hydrothermal cooking and moisture heat treatment
Our preliminary experiments showed that the critical gelation concentration of soy protein isolates under moisture heat treatment and hydrothermal cooking were 9% and 11%, respectively. Thus, 100 mL of SPI dispersions in distilled water (8%, w/v) were placed and sealed in 200 mL beakers. Then, the beakers were placed in a water bath at 90°C or in an autoclave at 121°C (0.1 MPa, gauge pressure) for 30 min. After moisture heat treatment or hydrothermal cooking, some water was added to the beakers, which was equal to the quantity of the evaporated one, followed by cooling by immersing in an ice bath, and then equilibrated to room temperature. The above solutions were then centrifuged at 10,000×g for 10 min, and supernatants were collected as soluble aggregates. 0.05% NaN3 was added to the supernatants, prior to storing at 4°C.
Measurement of mean hydrodynamic diameter
The mean hydrodynamic diameter of the proteins or aggregates in native and treated samples was measured by a dynamic light scattering technique using a Zetasizer Nano ZS (Malvern Instruments Ltd., Malvern, Worcestershire, UK) equipped with a 4 mW helium/neon laser at a wavelength output of 633 nm. Droplet sizing was performed at 25°C at 10-s intervals in a particle-sizing cell using backscattering technology. The intensity of light scattered from the proteins in the dispersions was used to calculate the mean hydrodynamic diameter, based on the Stokes-Einstein equation, assuming the proteins to be spherical. For each sample, the mean and standard deviation were calculated from at least five measurements. The native and treated-SPI dispersions were also filtered through 0.45 mm Millipore membrane prior to analysis. The protein concentration is 5 mg/mL.
Zeta potential (ζ) measurement
The ζ values of the proteins in SPI dispersions(native and treated), were measured by a laser doppler velocimetry and phase analysis light scattering (M3-PALS) technique using a Malvern Zetasizer Nano ZS (ZEN3600) instrument (Malvern Instruments Ltd., Malvern, Worcestershire, UK), in connection with a multipurpose autotitrator (model MPT-2, Malvern Instruments, Worcestershire, UK). The protein samples were prepared in deionized water, and diluted to 0.5% (w/v). The diluted protein dispersions were filtered with a 0.45 mm Millipore membrane, prior to the analysis. One milliliter of each diluted sample was put in an electrophoresis cell (Model DTS 1060C, Malvern Instruments Ltd., Malvern, and Worcestershire, UK). The temperature of the cell was maintained at ambient temperature. The data was the average values of three measurements performed with three individually prepared protein dispersions.
Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE)
SDS-PAGE experiments were performed according to the discontinuous buffer system of Laemmli [23] at 5% stacking gel and 12.5% separating gel using gel electrophoresis apparatus DYCZ-30 (Beijing LIUYI Instrument Factory, China). The protein samples were directly dissolved in the sample buffer, namely 0.125 M Tris-HCl buffer (pH 8.0) containing 1.0% (w/v) SDS, 0.05% (w/v) blue bromophenol, 30% (v/v) glycerol with or without 5% (v/v) 2-ME. Then the samples were vortexed for about 5 s, heated at 100°C for 5 min, and centrifuged (10,000×g, 10 min). The samples (containing about 5 µg protein for each) were loaded, and then the electrophoresis was run at 20 mA in the stacking gel and at 40 mA in separating gel until the tracking dye reached the bottom of the gel. Lastly, the gel was then dyed with Coomassie Blue R250 and destained. The band patterns were then photographed and analyzed with Quantity One software version 4.4 (Bio-Rad Laboratories Inc., USA). The relative quantity of each subunit (protein band) was calculated from its respective percent area on the densitograms against the areas of total subunits.
Gel Permeation Chromatography (GPC)
GPC analysis was performed on a Waters Breeze system equipped with a Water 1525 pump and Water 2487 UV detector (Waters, USA). The treated samples were diluted to 5 mg/mL by the use of 50 mM sodium phosphate (pH 7.2) containing 50 mM NaCl, centrifuged at 10000 g for 20 min, and filtered through 0.45 µm filters (Millipore, Fisher Sci.). The same buffer was used as mobile phase, and aliquots (20 µL) were injected into a prepacked TSK G4000SWxl column (TOSOH, Japan). The flow rate was 0.7 mL/min. The elution was monitored by absorbance at 280 nm. All data were collected and analyzed by Breeze software (Waters, Division of Millipore, and Milford, MA, USA). Bovine thyroid (669 kDa), rabbit muscle (158 kDa) and chicken egg white (75 kDa) obtained from GE Healthcare (Little Chalfont, Buckinghamshire, United Kingdom) were used as standard proteins for calibration.
Determination of the Free Sulfhydryl Group (SH) and Disulfide Bond (SS) contents
The SH and SS contents of samples were determined by the method of Beveridge et al. [24]. Protein samples (75 mg) were dissolved in 10 mL of Tris-Gly buffer (0.086 M Tris, 0.09 M glycine, and 0.04 M EDTA, pH 8.0) containing 8 M urea. The solution was gently stirred overnight until a homogeneous dispersion was achieved. For SH content determination, 4mL of the Tris-Gly buffer was added to 1 mL of protein solution. Then, 0.05 mL of Ellmanʼs reagent (5, 50-dithio-bis-2-nitrobenzoic acid in Tris-Gly buffer, 4 mg/mL) was added, and absorbance was measured at 412 nm after 5 min. For total SH content analysis, 0.05 mL of β-ME and 4 mL of Tris-Gly buffer were added to 1 mL of the protein solution. The mixture was incubated for 1 h at room temperature. After an additional hour of incubation with 10 mL of 12% TCA, the mixtures were centrifuged at 5000 g for 10 min. The precipitate was twice resuspended in 5 mL of 12% TCA and centrifuged to remove 2-ME. The precipitate was dissolved in 10 mL of Tris-Gly buffer. Then 0.04 mL of Ellmanʼs reagent was added to 4 mL of this protein solution, and the absorbance was measured at 412 nm after 5 min. The calculation was as follows: µM SH/g = 73.53×A412×D/C; where A412 is the absorbance at 412 nm, C is the sample concentration (mg/mL), D is the dilution factor, 5 and 10 are used for SH and total SH content analysis, respectively, and 73.53 is derived from 106/(1.36×104); 1.36×104 is the molar absorptivity [25], and 106 is for the conversion from molar basis to µM/mL basis and from mg solids to g solids. Half of the value after subtracting the SH value from the total SH value was defined as the SS content.
Far-UV and Near-UV Circular Dichroism (CD) Spectroscopy
Far-UV and near-UV CD spectra were obtained using an MOS-450 spectropolarimeter (Biologic Science Instrument, France). The far-UV CD spectroscopy measurements were performed in a quartz cuvette of 2 mm with a protein concentration around 0.1 mg/mL in 10 mM PBS (pH 7.0). The sample was scanned from 190 to 250 nm. The near-UV CD spectroscopy measurements were performed in a 1 cm quartz cuvette with a protein concentration around 1.0 mg/mL. The sample was scanned over a wavelength range from 250 to 320 nm. For both measurements, the spectra were an average of eight scans. The following parameters were used: step resolution, 1 nm; acquisition duration, 1 s; bandwidth, 0.5 nm; sensitivity, 100 mdeg. The cell was thermostatted with a Peltier element at 25°C, unless specified otherwise. The concentration of the proteins was determined by the Lowry method [26] using BSA as the standard. Recorded spectra were corrected by subtraction of the spectrum of a proteinfree buffer. A mean value of 112 for the amino acid residue was assumed in all calculations and CD measurements expressed as the mean residue ellipticity (θ) in deg.cm-2.dmol-1. The secondary structure compositions of the samples were estimated from the far-UV CD spectra using the CONTIN/LL program in CDPro software, using 43 kinds of soluble proteins as the reference set [27,28]. Each data point was the mean of duplicate measurements.
Intrinsic fluorescence spectroscopy and surface hydrophobicity (H0)
Intrinsic emission fluorescence spectra of samples were determined in a RF-5301 PC fluorophotometer (Shimadzu Co., Kyoto, Japan). Protein dispersions (0.15 mg/mL) were prepared in 10 mM phosphate buffer (pH 7.0). Protein solutions were excited at 295 nm, and emission spectra were recorded from 300 to 400 nm at a constant slit of 5 nm for both excitation and emission. H0 was determined using ANS-, according to the method in pervious study [29]. In brief, stock solutions of 8×10-3 M ANS-, and 1.5% (w/v) protein dispersions were prepared in 10 mM phosphate buffer (pH 7.0). To successive samples containing 4 mL of buffer and 20 µL of ANS- stock solutions were added 10, 20, 30, 40, and 50 µL of 1.5% protein solution, respectively. Samples were mixed in vortex mixer for about 5 s. Fluorescence intensity (FI) was measured at wavelengths of 390 nm (excitation) and 470 nm (emission) at 20±0.5°C using a RF-5301 PC fluorophotometer (Shimadzu Co., Kyoto, Japan), with a constant excitation and emission slit of 5 nm. The FI for each sample was then computed by subtracting the FI attributed to protein in buffer. The initial slope of the FI versus protein concentration (mg/mL) plot (calculated by linear regression analysis) was used as an index of surface hydrophobicity (H0).
Ultraviolet (UV) Spectroscopy
Samples were dissolved in 10 mM sodium phosphate (pH 7.2) and centrifuged at 10,000 g for 20 min. The protein concentrations of all samples were adjusted to 0.3 mg/mL. Baseline corrected UV spectra were recorded between 250 and 300 nm at medium speed in the double beam mode using a UV2300 spectrophotometer (Techcomp, China). The data interval was 0.1 nm. Second-derivative UV spectra were analyzed using Origin 8.0 software (Origin-Lab Corp., Northampton, MA).
Results and Discussion
Physicochemical properties
Mean hydrodynamic diameters by DLS and ζ-potentials
The thermal aggregation of the proteins in SPI dispersions (at pH 7.0) was evaluated by dynamic light scattering technique, and the mean hydrodynamic diameters of the protein aggregates formed are shown in figure 1. The mean hydrodynamic diameter of the proteins in untreated SPI dispersion (designated as N-SPI, served as control) was about 40 nm. As expected, the mean hydrodynamic diameter of the proteins in modified SPI dispersions treated by hydrothermal cooking (designated as HC-SPI) and moisture heat treatment (designated as HT-SPI) remarkably increased, indicating thermal aggregation of the proteins. Significantly higher value of mean hydrodynamic diameter of HC-SPI (about 325 nm), as compared to HT-SPI (about 200 nm) Figure 1 indicated much higher extent of protein aggregation in HC-SPI. This is well consistent with the turbidity data of the proteins (data not shown), suggesting that hydrothermal cooking, which included steam treatment with high temperature(121°C) and gauge pressure(0.1 MPa), was more favorable for aggregation of the proteins than heat treatment(especially 90°C in our study).
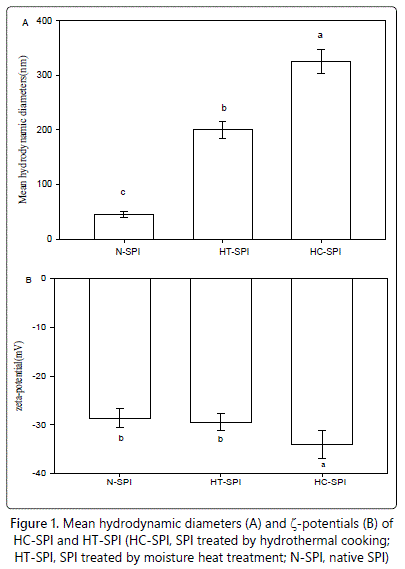
To confirm the changes in electrostatic forces, we determined zeta potentials (ζ) of the proteins in native and treated SPI, as shown in figure 1. ζ potential is commonly used in the interpretation of protein suspension stability [30]. The native SPI (N-SPI) showed its ζ value around -28 mV, which was less than that reported in former study [31]. The difference in ζ values may be attributed to the test conditions and materials. After moisture heat treatment, the ζ was almost unchanged. However, the ζ value of HC-SPI (about-33 mV) was slightly but significantly larger than that of HT-SPI. The reason may be due to the lowering pH value of about 0.4 (data not shown) of HC-SPI comparing with N-SPI, which resulted in more protonation of carboxyl groups and de-protonation of amino groups of the proteins, although the mechanism in pH-lowering of SPI treated by hydrothermal cooking was unclear. As for the induced aggregates, the different ζ potential values may also be related to their morphology to some extent, since the external morphological appearance of aggregates are highly sensitive to the amount of charge on the aggregating protein molecule [32].
SDS-PAGE profiles and GPC analysis
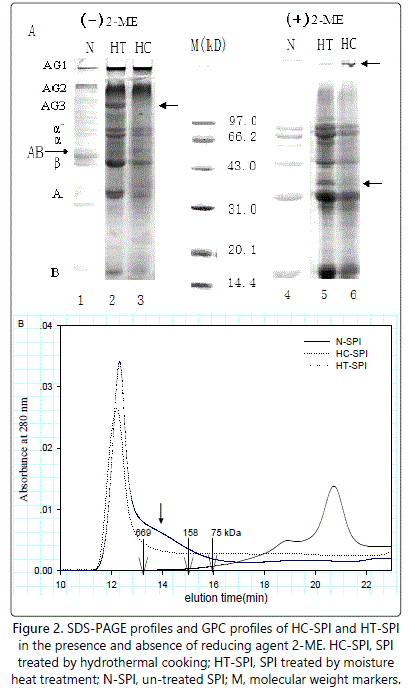
As expected, the native soy protein isolates showed typical SDS-PAGE profiles of β-conglycinin constituent subunits (α,α' and β) and glycinin constituent subunits (acidic and basic ones) (Figure 2A, lane 4). In the absence of 2-ME (Figure 2A), the N-SPI (Figure 2A, lane 1) showed a large band with Mr of 55 kDa attributed to AB band, and several bands with Mr ranging from 30 to over 100 kDa. The addition of 2-ME led to the dissociation of some bands in the control, such as AB band and some aggregated protein bands with high molecular weight, and the intensity of the constituent subunits of soy protein increased in the presence of 2-ME concomitantly (Figure 2A, lane 4), indicating that disulfide bonds play key roles in native SPI.
Figure 2A also shows the SDS-PAGE patterns of HC-SPI and HT-SPI. Both moisture heat treatment and hydrothermal cooking caused disappearance of the AB band at Mr of 55 kDa, and one band with Mr of 35 kDa and bands with lower molecular weights generated. (Figure 2A, lane 2 and lane 3). Meanwhile, several aggregates bands(designated as AG1, AG2 and AG3) were observed for HT-SPI (Figure 2A, lane2), while only AG1 and AG2 for HC-SPI (Figure 2A, lane3). In the presence of 2-ME, A3, with Mr of about 40 kDa, still existed in aggregate-form in the resultant aggregates corresponding to AG3 in the case of HC-SPI (Figure 2A, lane 6), suggesting that some compact structures (not accessible to SDS or/and 2-ME) may have existed in the resultant aggregates.
GPC was also performed to understand the aggregation behavior of the modified proteins by hydrothermal cooking and moisture heat treatment. The elution profiles of various samples are shown in figure 2B. The elution profiles of N-SPI showed major eluting peaks appearing at about 19 and 21 min, which attributed to β-conglycinin and glycinin, respectively. The molecular weights of the components were lower 158 kDa. In contrast, large aggregates were both observed for the treated-SPIs, as evidenced by the appearance of peaks eluting at 12.5 min and concomitant and significant decrease arround the typical peak areas (at 19 and 21 min). The average weights of heat-treated SPIs were over 669 kDa. It should be pointed out that the total peak area eluting at 12.5 min of HC-SPI was considerably smaller than that of HT-SPI, which seemed to be in contrary with the truth that hydrothermal cooking mediated a much higher extent of aggregation than moisture heat treatment, as evidenced by the significantly higher mean hydrodynamic diameters of HCSPI (Figure 1A). In fact, the additional filtration through 0.45 µm filters before GPC analysis may be a rational explanation for this decrease. Besides, a shoulder peak close to the peak at around 12.5 min, indicative of relative smaller aggregates, were observed only for HT-SPI, which may correspond to AG3 in the SDS-PAGE profiles (Figure 2A, lane 2). The above results further convinced that hydrothermal cooking was a stronger means to induce aggregation comparing with moisture heat treatment in our study.
Surface Hydrophobicity and ANS-Fluorescence Spectra
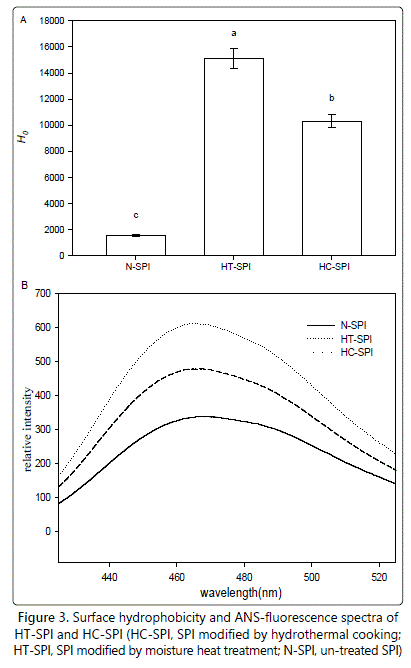
The surface hydrophobicity of native and treated SPI is presented in figure 3A. The surface hydrophobicity of native SPI was about 1500 (Figure 3A). However, the surface hydrophobicity of treated-SPI by moisture heat treatment or hydrothermal cooking rose dramatically to about 10000 and 15000, respectively, suggesting that lots of hydrophobic clusters initially distributed in the inner protein molecules were exposed to the molecule surface.
The fluorescence emission spectra of ANS (a polarity-sensitive fluorescent probe) upon binding to the proteins in HT-SPI and HC-SPI are shown in figure 3B. The maximum emission (λmax) of the ANS-probes upon complexation by the proteins in control was 467 nm. The λmax of ANS- in 10 mmolL-1 phosphate buffer (pH 7.0) appeared at 509 nm according to Haskard and Li-Chan [29]. The observed blue shift in λmax of ANS- probes upon complexation by the control (N-SPI) is indicative of an increase in hydrophobicity of the ANS-microenvironment upon protein binding. The maximum emission (λmax) of the ANS-probes upon complexation by the proteins in HT-SPI and HC-SPI was 466 and 464 nm, respectively. The larger blue shift in λmax in the case of HT-SPI, as compared to the case of HC-SPI, suggested a larger increase in hydrophobicity of the ANS- microenvironment upon protein binding. This is in good agreement with H0 (Figure 3A), indicating that more initially buried hydrophobic groups were exposed and induced by moisture heat treatment than hydrothermal cooking. A reasonable explanation is that, the high aggregation degree of HC-SPI led to a hydrophobic buried effect to some extent for several hydrophobic groups, initially exposed on the surface of the treated-SPI during hydrothermal cooking processing, thus resulting in smaller hydrophobicity (relative to that of HT-SPI). The above results also confirms the fact that, ANS binds expose hydrophobic surfaces in partially unfolded proteins with much higher affinity than native or completely unfolded proteins [32].
Total free sulfhydryl contents (SH) and disulfide bond (SS) contents
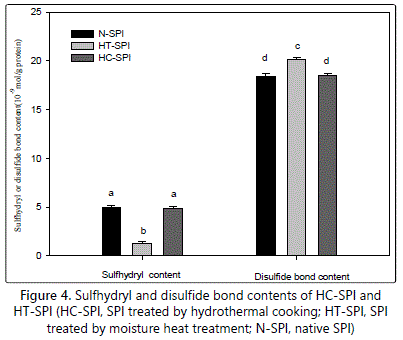
Figure 4 shows total free SH group contents and disulfide bond (SS) contents of native SPI and treated-SPI. In the native SPI, total free SH contents and disulfide bond (SS) contents achieved about 5.0 µmol/g and 18.4 µmol/g protein, respectively, which is consistent with former studies [34,35]. The free SH groups and disulfide bond (SS) contents may mainly attribute to glycinin in SPI, due to its much higher sulfhydryl group (approximately 6.3 µmol of free SH group/g of protein, assuming MW 320000) and disulfide bond (SS) contents as compared to β-conglycinin [36]. Moisture heat treatment resulted in significant decrease in total free SH contents and concomitant increase in disulfide bond (SS) contents (1.3 µmol/g proteins and about 20.1 µmol/g proteins, respectively)(Figure 4). Similar results had been observed in some other studies, taking other initial protein concentrations [37]. Unexpectedly, the total free SH content and disulfide bond (SS) contents of HC-SPI were almost unchanged as compared to N-SPI, with the value being 4.85 µmol/g proteins and 18.51 µmol/g proteins, respectively. SS bonds have been proved to play key roles both () HC-SPI and HT-SPI, indicated by SDS-PAGE profiles (Figure 2A, lane 3 and lane 6). Thus, discrepancy of SS bonds formation as for HC-SPI and HT-SPI may exist. The decrease of total free SH content in moisture heat treatment case was attributed to oxidation of the sulfhydryl groups or/and SH/SS interchange [34]. On the contrary, hydrothermal cooking in an autoclave system favored to disruption of all SS bonds followed by the rearrangement of SS/SH bonds through oxidation of the adjacent sulfhydryl groups [20]. Then, SS/SH bond rearrangement may occur in interprotein or/and intraprotein [20].
Conformational properties
Far-UV and Near-UV CD analysis
Secondary structures of proteins such as α-helices, β-sheets, β-turns and random coil can be characterized by far-UV CD spectra in the wavelength range from 190 to 260 nm [38]. As shown in figure 5A, the far-UV CD spectrum for N-SPI showed a strong positive peak in the vicinity of 197 nm, and a broad shoulder that extends from 207 to 230 nm. The spectrum results indicated that N-SPI is with a highly ordered structure of β-structure [39]. When the N-SPI subjected to moisture heat treatment or hydrothermal cooking, the positive peak around 197 nm and the negative peak at about 215 nm decreased, suggesting heat treatment increase the structural flexibility. The composition of the secondary structures is also presented in table 1. The secondary structures of HT-SPI and HC-SPI comprised mainly β-sheets, which significantly decreased α-helix contents but increased random coil structures as compared to N-SPI. The random coil content of HC-SPI increased more than that of HT-SPI. This result implied that the conformational changes of HC-SPI aggregates were more disordered due to the process of higher temperature and pressure.
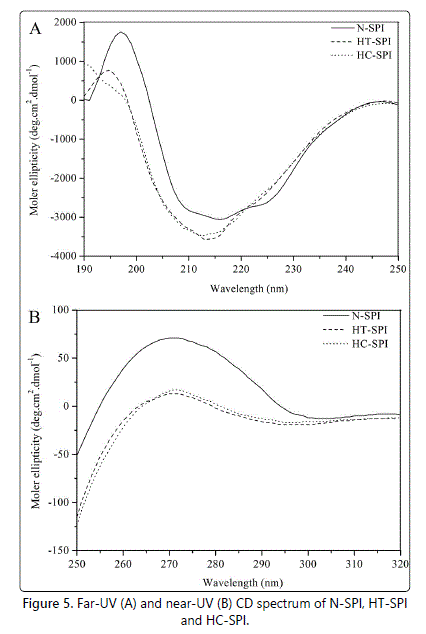
The tertiary conformations of HT-SPI and HC-SPI were analyzed by near-UV CD spectroscopic technique. The actual shape and magnitude of the near-UV CD spectrum of a protein will depend on the number of each type of aromatic amino acid present, their mobility, the nature of their environment (H-bonding, polar groups and polarisability), as well as their spatial disposition in the protein [40]. Figure 5B shows the near-UV CD spectra of N-SPI, HT-SPI and HC-SPI at pH 7.0. Compared to N-SPI, HT-SPI and HC-SPI exhibited significantly different patterns of CD spectra in the near-UV region, indicating that substantial differences existed in the arrangement/proximity and exposure of aromatic amino acids on the protein surfaces after moisture heat treatment or hydrothermal cooking. The decreases in ellipticity suggested that an increasing number of aromatic side-chains were exposed to a modified environment, and these changes are related to the loss of native-like structure.

Intrinsic Emission Fluorescence spectra and Second-derivative UV spectra
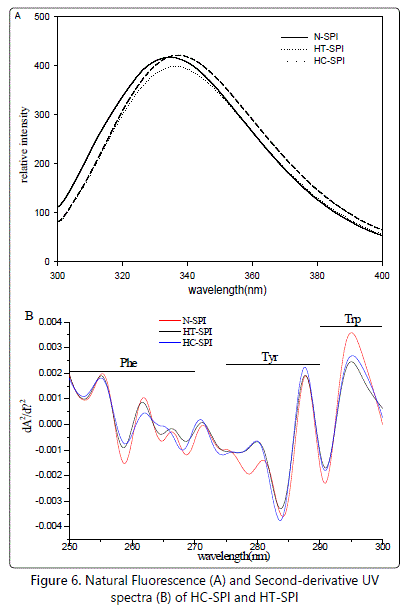
The intrinsic fluorescence spectrum is determined chiefly by the polarity of the environment of the tryptophan (Trp) residues and provided a sensitive means of monitoring the conformational changes in proteins and protein-protein as well as ligand-protein interactions [33]. The difference in the hydrophobic clusters on the molecular surfaces of the proteins in HT-SPI and HC-SPI could also be well reflected in intrinsic fluorescence spectral analysis Figure 6A. The emission fluorescence spectrum is determined chiefly by the polarity of the environment of the tryptophan and by their specific interactions and provides a sensitive means of characterizing proteins and their conformation, since the fluorescence emission maximum suffers a red shift when chromophores become more exposed to solvent [33]. The proteins in N-SPI showed a typical intrinsic fluorescence spectrum, with an emission maximum (λmax) at around 3 (Lange and Balny, 2002). The changes in amplitude, described 35 nm. This is a characteristic fluorescence profile of tryptophan residues in a relatively hydrophobic environment, such as the interior of a globulin [41]. In contrast, the λmax in HT-SPI and HC-SPI were about 336.5 and 338 nm, respectively. The observed red shift indicated that the Trp residues in HT-SPI were less exposed to the polar environment than that in HC-SPI.
The major components of SPI, β-Conglycinin and glycinin contain all types of aromatic acid residues, such as Phe, Tyr and Trp. The second-derivative UV spectroscopy was thereby selected to critically delineate the distribution of aromatic acid residues in modified proteins, such as HT-SPI and HC-SPI. Figure 6B shows the second-derivative UV spectra of native and modified SPI. The position and amplitude of the derivative spectral bands can be used as sensitive probes to characterize the polarity of microenvironments. Generally, a decrease of the solvent polarity results in red-shifts of the derivative bands [42]. The changes in amplitude, described by calculating the ratio (r = a/b) of the two peak-to-trough values marked in figure 6B, can also be used to access the environments of the Tyr residues [42]. Compared with native SPI, treated SPI showed distinct distributions among aromatic acid residues (Phe, Tyr and Trp), as shown in figure 6B.
The slight blue-shifts of bands in the Phe residue region (250-270 nm) occurred for HT-SPI Figure 6B indicating a gradual transition to more polar environments. The UV spectra of HT-SPI showed the continuous blue-shifts of bands in the Tyr residue region (270-285 nm). In the case of HC-SPI, significantly larger blue-shifts of bands corresponding to Tyr residues were observed comparing with HT-SPI, suggesting that Tyr residue microenvironments in HC-SPI were more exposed to polar environment. The ratio (r=a/b) of the two peak-to-trough values further confirmed the results.
Conclusion
In conclusion, the physicochemical and conformational properties, including surface net charge, hydrophobicity, free SH contents and SS bond contents, as well as secondary and tertiary conformations of soy protein isolates were modified by hydrothermal cooking and moisture heat treatment, as compared to native soy protein isolates. SS bonds both play key roles in aggregates formation of SPI induced by hydrothermal cooking and moisture heat treatment. SH/SS interchange reactions were more predominant in the case of hydrothermal cooking. The secondary structure of soy protein isolates modified by hydrothermal cooking were much more unordered than heat treatment, while their tertiary and quaternary conformations showed varying exposure extent with varying aromatic acid residues (Phe, Tyr and Trp). These results can thus provide useful suggestions for modification of soy protein isolates with great functionality using hydrothermal cooking or/and moisture heat treatment.
Conflict of interest: The authors declare that there are no conflicts of interest.
References
- Iwabuchi S, Yamauchi F. Determination of glycinin and β-conglycinin in soybean proteins by immunological methods. J Agric Food Chem. 1987; 35(2): 200-205. doi: 10.1021/jf00074a009
- Iwabuchi S, Yamauchi F. Electrophoretic analysis of whey proteins presents in soybean globulin fractions. J Agric Food Chem. 1987; 35(2): 205-209. doi: 10.1021/jf00074a010
- Kinsella JE. Functional properties of soy proteins. J Am Oil Chem Soc. 1979; 56(3): 242-258. doi: 10.1007/BF02671468
- Yamauchi F, Yamagishi T, Iwabuchi S. Molecular understanding of heat induced phenomena of soybean protein. Food Res Int. 1991; 7(3): 283-322. doi: 10.1080/87559129109540914
- Hermansson AM. Physico-chemical aspects of soy proteins structure formation. J Texture Stud. 1978; 9(1-2): 33-58. doi: 10.1111/j.1745-4603.1978.tb01293.x
- Petruccelli S, Añón MC. The relationship between the method of obtention and the structural and functional properties of soy protein isolates. 1. Structural and hydration properties. J Agric Food Chem. 1994; 42(10): 2161-2169. doi: 10.1021/jf00046a018
- Petruccelli S, Añón MC. The relationship between the method of obtention and the structural and functional properties of soy protein isolates. 2. Surface properties. J Agric Food Chem. 1994; 42(10): 2170-2176. doi: 10.1021/jf00046a018
- Dio E. Gels and gelling of globular proteins. Trends Food Sci Technol. 1993; 4(1): 1-5. doi: 10.1016/S0924-2244(05)80003-2
- Chiti F, Dobson CM. Amyloid formation by globular proteins under native conditions. Nat Chem Biol. 2009; 5(1): 15-22. doi: 10.1038/nchembio.131
- Keerati-u-rai M, Correding M. Heat-induced changes in oil-in-water emulsions stabilized with soy protein isolate. Food Hydrocoll. 2009; 23(8): 2141-2148. doi: 10.1016/j.foodhyd.2009.05.010
- Utsumi S, Damodaran S, Kinsella JE. Heat-induced interactions between soybean proteins: Preferential association of 11S basic subunits and β subunits of 7S. J Agric Food Chem. 1984; 32(6): 1406-1412. doi: 10.1021/jf00126a047
- Wagner JR, Añón MC. Influence of denaturation degree, hydrophobicity and sulphydryl content on solubility and water absorbing capacity of soy protein isolates. J Food Sci. 1990; 50(3): 765-770. doi: 10.1111/j.1365-2621.1990.tb05225.x
- Arrese EL, Sorgentini DA, Wagner JR and Añón MC. Electrophoretic, solubility, and functional properties of commercial soy protein isolates. J Agric Food Chem. 1991; 39(6): 1029-1032. doi: 10.1021/jf00006a004
- German B, Damodaran S, Kinsella JE. Thermal dissociation and association behavior of soy proteins. J Agric Food Chem. 1982; 30(5): 807-811. doi: 10.1021/jf00113a002
- Damodaran S, Kinsella JE. Effect of conglycinin on the thermal aggregation of glycinin. J Agric Food Chem. 1982; 30(5): 812-817. doi: 10.1021/jf00113a003
- Petruccelli S, Añón MC. Thermal aggregation of soy protein isolates. J Agric Food Chem. 1995; 43(12): 3035-3041. doi: 10.1021/jf00060a009
- Sorgentini DA, Wagner JR, Anon MC. Effects of thermal treatment of soy protein isolate on the characteristics and structure-function relationship of soluble and insoluble fractions. J Agric Food Chem. 1995; 43(9): 2471-2479. doi: 10.1021/jf00057a029
- Wang C, Johnson LA. Functional properties of hydrothermally cooked soy protein products. J Am Oil Chem Soc. 2000; 78(2): 189-195. doi: 10.1007/s11746-001-0242-y
- Wang H, Johnson LA. Refunctionalization of extruded-expelled soybean meals. J Am Oil Chem Soc. 2004; 81(8): 789-794. doi: 10.1007/s11746-004-0979-3
- Chinnaswamy R, Bassi S, Maningat CC. Process for preparing hybrid proteins. 2004; US 0086613 A1.
- Zheng HG, Yang XQ, Tang CH, Li L, Ahmad I. Preparation of soluble soybean protein aggregates (SSPA) from insoluble soybean protein concentrates (SPC) and its functional properties. Food Res Int. 2008; 41(2): 154-164. doi: 10.1016/j.foodres.2007.10.013
- Wu W, Hettiarachchy NS, Kalapathy U, Williams WP. Functional properties and nutritional quality of alkali and heat-treated soy protein isolate. J Food Qual. 1999; 22(2): 119-133. doi: 10.1111/j.1745-4557.1999.tb00545.x
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970; 227: 680-685. doi: 10.1038/227680a0
- Beveridge T, Toma SJ, Nakai S. Determination of SH and S-S groups in some food proteins using Ellman's reagent. J Food Sci. 1974; 39(1): 49-51. doi: 10.1111/j.1365-2621.1974.tb00984.x
- Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys. 1959; 82(1): 70-72.
- Lowry OH, Rosembroug HJ, Lewis A, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. 1951; 193: 265-275.
- Sreerama N, Woody RW. Estimation of protein secondary structure from circular dichroism spectra: Comparison of CONTIN, SELCON and CDSSTR methods with an expanded reference set. Anal Biochem. 2000; 287(2): 252-260.
- Yang JT, Wu CS, Martinez HM. Calculation of protein conformation from circular dichroism. Methods Enzymol. 1986; 130: 208-269.
- Haskard CA, Li-Chan ECY. Hydrophobicity of bovine serum albumin and ovalbumin determined using uncharged (PRODAN) and anionic (ANS-) fluorescent probes. J Agric Food Chem. 1998; 46(7): 2671-2677. doi: 10.1021/jf970876y
- Jachimska B, Wasilewska M, Adamczyk Z. Characterization of globular protein solutions by dynamic light scattering, electrophoretic mobility, and viscosity measurements. Langmuir. 2008; 24(13): 6866-6872. doi: 10.1021/la800548p
- Malhotra A, Coupland JN. The effect of surfactants on the solubility, zeta potential, and viscosity of soy protein isolates. Food Hydrocoll. 2004; 18(1): 101-108. doi: 10.1016/S0268-005X(03)00047-X
- Krebs MR, Devlin GL, Donald AM. Amyloid fibril-like structure underlies the aggregate structure across the pH range for ?-Lactoglobulin. Biophys J. 2009; 96(12): 5013-5019. doi: 10.1016/j.bpj.2009.03.028
- Pallarès I, Vendrell J, Avilès FX, Ventura S. Amyloid fibril formation by a partially structured intermediate state of a-chymotrypsin. J Mol Biol. 2004; 342(1): 321-31.
- Shimada K, Cheftel JC. Determination of sulfhydryl groups and disulfide bonds in heat-induced gels of soy protein isolate. J Agric Food Chem.1988; 36(1): 147-153. doi: 10.1021/jf00079a038
- Tang CH. Thermal denaturation and gelation of vicilin-rich protein isolates from three Phaseolus legumes: A comparative study. LWT-Food Science and Technology. 2008; 41(8): 1380-1388. doi: 10.1016/j.lwt.2007.08.025
- Draper M, Catsimpoolas N. Disulfide and sulfhydryl groups in glycinin. Cereal Chem. 1978; 55: 16-22.
- Hashizume K, Watanabe T. Influence of heating temperature on conformational changes of soybean proteins. Agric Biol Chem. 1979; 43(4): 683-690. doi: 10.1080/00021369.1979.10863529
- Kelly SM, Price NC. The application of circular dichroism to studies of protein folding and unfolding. Biochim Biophys Acta. 1997; 1338(2): 161-185.
- Zirwer D, Gast K, Welfle H, Schlesier B,Schwenke KD. Secondary structure of globulins from plant seeds: A re-evaluation from circular dichroism measurements. Int J Biol Macromol.1985; 7(2): 105-108. doi: 10.1016/0141-8130(85)90039-X
- Kelly SM, Jess TJ, Price NC. How to study proteins by circular dichroism. Biochim Biophys Acta. 2005; 1751(2): 119-139.
- Dufour E, Hoa GH, Haertlé, T. High-pressure effects of ͎-lactoglobulin interactions with ligands studied by fluorescence. Biochim Biophys Acta. 1994; 1206(2): 166-172.
- Lange R, Balny C. UV-visible derivative spectroscopy under high pressure. Biochim Biophys Acta. 2002; 1595(1-2): 80-93. doi: 10.1016/S0167-4838(01)00336-3
