


Case Report
Ichthyosis Follicularis, Alopecia and Photophobia Syndrome-A Rare Case in Female
MGM Medical College, Aurangabad, Maharashtra, India
*Corresponding author: Dr. Pranali Uttam Rathod, Junior Resident, MGM Medical College, Aurangabad, Maharashtra, India, Tel: +91 8888 4451 27, E-mail: dr.pranalirathod@gmail.com
Received: March 29, 2018 Accepted: July 30, 2018 Published: August 3, 2018
Citation: Gulanikar AD, Rathod PU, Atram V. Ichthyosis Follicularis, Alopecia and Photophobia Syndrome-A Rare Case in Female. Madridge J Dermatol Res. 2018; 3(2): 68-69. doi: 10.18689/mjdr-1000115
Copyright: © 2018 The Author(s). This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Icthyosis Follicularis, Alopecia and Photophobia (IFAP) syndrome is genetically inherited syndrome characterized by Ichthyosis follicularis, alopecia & photophobia. It is X linked recessive inheritance trait and seen almost exclusively in male. It is characterize as spiny follicular papules all over body since birth.
Congenital atrichia, photophobia are most prominent feature in this syndrome. We are presenting a rare case of IFAP Syndrome of a nine-year old female patient. Only 40 cases of IFAP syndrome reported worldwide.
Keywords: Ichthyosis Follicularis; Alopecia; Photophobia; Spiny Follicular Papules.
Introduction
The IFAP syndrome is a rare X-linked genetic disorder and reported rarely. Only fourty cases of IFAP syndrome reported till date worldwide [1]. It is characterized by the triad of Ichthyosis, Follicularis, Alopecia, and Photophobia from birth. Other features such as short stature, intellectual disability, and seizures may develop in the first few years of life. Two kinds of dermatological abnormalities have been described in IFAP syndrome. The ‘classical' IFAP syndrome is characterized as the presence of generalized cutaneous thorn like projections, giving a ‘nutmeg grater' or ‘prickly surface 'or a ‘rose leaf' feeling and second characterized collodian membrane (occasionally present). Ichthyosis and alopecia reported in all patients. Atopic eczema like lesion, angular chelitis, nails dystrophy reported in most of cases. IFAP is sometimes accompanied by short stature, intellectual disability and seizures. Photophobia may also be present since the first year of life or appears in infancy or early childhood. Mode of inheritance is X-linked recessive, thus mostly affecting males. Affected or carrier females may manifest some of its clinical features.
Case Report
A 9-year-old female born by non consanguineous marriage presented with dry rough skin & loss of hair since age of four years. There was history of photophobia, redness, watering of eyes since birth. She had undergone cataract surgery one year back. Her growth & developmental milestones were normal. No sweating defect, hearing deficit, nail dystrophy. No systemic involvement. No similar complaint in family members.
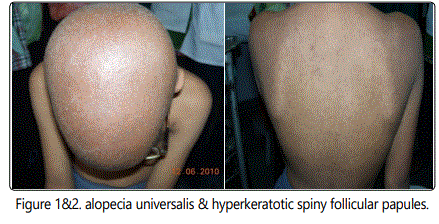
On dermatological examination - hyperkeratotic spiny follicular papules having sand paper like texture of the skin (fig 2) & alopecia universalis (fig 1). There was no erythema and scarring. Teeth, genital and oral mucosa were in normal limits. General examination was within normal limits. Ocular examination showed photophobia, myopia, left eye aphakia, and conjunctival congestion. Slit lamp examination showed diffused corneal vascularization and fundoscopy within normal limits.
Biopsy specimens from the scalp in serial sections showed keratin plugs occupying dilated hair follicles and hyperkeratosis of the skin. In the dermis follicles were in telogen phase. Sweat glands and a few sebaceous glands were present.
Discussion
IFAP syndrome is type of keratinization disorder. It results from marked defect in pilosebaceous apparatus. This disorder results from mutations in the MBTPS2 gene that impairs cholesterol homeostasis and the ability to cope with endoplasmic reticulum stress. Males are having severe manifestations indicating X-linked recessive pattern of inheritance. The risk for a female carrier to have an affected son is 50% due to Lyon effect of X - inactivation. The female carriers may show linear areas of follicular hyperkeratosis as well as patchy linear hairlessness. Our female patient had generalized skin involvement indicating autosomal dominant mode of transmission.The mutation might also arise in the patient de novo [2]. IFAP syndrome cannot be detected prenatally by ultrasonography. If the mutation has been previously detected in the family, prenatal diagnosis is advised to female carriers.
Ocular manifestations of IFAP syndrome are: photophobia, corneal scars, punctate keratopathy, corneal erosion and neovascularization, and atopic keratoconjunctivitis. Chronic tearing, cataract, horizontal nystagmus, astigmatism and myopia.
Neurological manifestations are: global developmental delay, seizures, and mild inner cerebral atrophy. Other clinical features are short stature, olivo-cerebellar atrophy, malformation of the temporal lobes, and hypoplasia of the corpus callosum have been reported.
Dysmorphic features such as frontal bossing, choanal atresia, and large ears. Intestinal anomalies such as omphalocele, Hirschsprung's disease, congenital aganglionic mega colon, stenosis of the small intestine, renal, vertebral, and testicular anomalies, inguinal hernia, cleft hands, and recurrent infections [3].
Management is difficult and symptomatic treatment is preferred. A moderate response seen to acitretin therapy at a dose of 0.3 to 1mg/kg/day. Improvement is seen in cutaneous features and corneal erosions. No improvement seen in alopecia and photophobia [2, 4]. Follicular hyperkeratosis can be treated using topical keratolytics, urea preparations, and emollients. Topical retinoid are not used because of their irritation potential. Corneal vascularization is relentless in affected boys and does not respond to topical corticosteroid therapy. Intensive lubrication of the ocular surface remains the mainstay of therapy [5]. Seizures, if any must be treated accordingly. Life expectancy varies from death in the neonatal period to normal surviving. The oldest reported patient was 33 years old [6]. Cardio-pulmonary complications are the main cause of death. In our case based on history & clinical examination IFAP syndrome was diagnosed. It is rarely seen in females. Dermatological manifestations in females are minimal with localized follicular lesions and localized hair loss. In our case extensive skin lesions, alopecia universalis and photophobia all the features which are most commonly seen in male patients were present.
Extensive involvement in female as in our case may suggest autosomal dominant mode of transmission could be present.

The genetic studies in support of our diagnosis are lacking but classic clinical presentation has made our diagnosis easier. Our next step is to get genetic studies done to detect possible mode of transmission.
The differential diagnosis considered are dermotrichic syndrome, keratitis-ichthyosis-deafness syndrome (KID), keratosis follicularisspinulosadecalvans (KFSD), hereditarymucoepithelial dysplasia (HMD) and can be differentiated from IFAP syndrome on clinical examination.
Conflict of Interest
The authors declare no conflicts of interest.
References
Contact us for any additional information - contact@madridge.org
 Madridge Publishers is licensed under a Creative Commons Attribution 4.0 International License.
Madridge Publishers is licensed under a Creative Commons Attribution 4.0 International License.