


Case Report
Cutaneous Plasmacytosis treated with Tocilizumab: A Case Report and Review of the Literature
Division of Dermatology, City of Hope Comprehensive Cancer Center, USA
*Corresponding author: Jae Yeon Jung, Division of Dermatology, City of Hope Comprehensive Cancer Center, 1500 East Duarte Road, Duarte, CA 91010, USA, Tel: 626-256-4673, ext: 65621, Fax: 626-301-8341, E-mail: jjung@coh.org
Received: August 5, 2016 Accepted: August 31, 2016 Published: September 3, 2016
Citation: Almazan TH, Jung JY. Cutaneous Plasmacytosis treated with Tocilizumab: A Case Report and Review of the Literature. Madridge J Dermatol Res. 2016; 1(1): 1-7. doi: 10.18689/mjdr-1000101
Copyright: © 2016 The Author(s). This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Objective: To review the current literature relevant to cutaneous plasmacytosis in both children and adults, with a focus on treatments documented as efficacious. Differing drug classes used to treat this disease and their respective outcomes will also be reviewed.
Data sources: A pubmed.org search was conducted using the search phrase "cutaneous plasmacytosis."
Data extraction: Information regarding clinical presentation, treatment, and pathophysiology of cutaneous plasmacytosis was specifically identified and reviewed.
Conclusions: Cutaneous and systemic plasmacytosis can affect both children and adults and presently lacks consensus statements on definitive treatment regimens. A continued consolidation of the little knowledge we have about this disease is needed, in order to uncover possible patterns of treatment efficacy, and to maintain awareness of this unusual disease in a dermatologist's differential diagnoses.
Keywords: Cutaneous Plasmacytosis; Tocilizumab; Lymphoplasmacytic Disorder; Myeloma cells; Plasma cells; Filipino woman.
Introduction
Cutaneous and systemic plasmacytosis is an extraordinarily rare, reactive lymphoplasmacytic disorder characterized by mature plasma cell proliferation in the skin or organs [1,2]. Unlike multiple myeloma, the plasma cells in plasmacytosis do not showcase atypia and are therefore considered benign. In the majority of cases, the expansion of plasma cells is polyclonal [3]. The vast majority of cases have been described in middle-aged individuals of Japanese decent. To our knowledge, only one other case has been described in a Filipino individual [4-6]. Here, we describe a second case of plasmacytosis in a Filipino woman.
Case Report
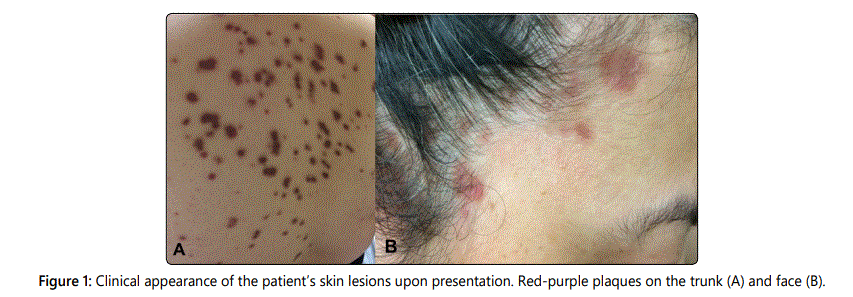
A 53-year-old Filipino woman presented to an outpatient clinic with numerous red-brown plaques on her face and trunk. The lesions were non-painful and had been present for approximately four years. She denied any constitutional symptoms including fevers, weight loss, or night sweats. An excisional biopsy of a chest wall lesion demonstrated mild, superficial perivascular plasma cells and anetoderma. A second biopsy of a similar lesion on the forehead revealed a possible diagnosis of cutaneous follicular lymphoma. Appropriately, the patient received an expedited bone marrow biopsy that resulted as negative for malignancy. A PET/CT scan uncovered FDG-avid bilateral cervical lymph node enlargement, along with minimally FDG-avid axillary, mediastinal, iliac, and inguinal adenopathy. An ultrasound-guided core biopsy of a cervical lymph node did not exhibit any evidence of lymphoma.
An excisional biopsy of a palpable groin lymph node disclosed reactive follicular hyperplasia, marked plasmacytosis, and Castleman-like features. The patient was subsequently started on thalidomide and prednisone to treat presumptive Castleman's disease. Despite therapy, she noted progression of her skin lesions, with additional hyperpigmented patches having evolved on her right breast and ankle.
Laboratory tests were significant for an elevated white blood cell count of 14.7 K/μL and an increased monocyte differential of 16%. The patient exhibited a normocytic anemia (hemoglobin of 11.1 g/dL) along with mild thrombocytosis (platelets 502 K/μL). Serum electrolytes were within normal limits. Total protein was elevated at 9.6 g/dL. Serum protein electrophoresis revealed an elevated gamma fraction without a monoclonal spike. No paraprotein was identified by immunofixation. Serum IgG was elevated at 2180 (normal range 700-1600 mg/dL). Additionally, serum IL-6 was elevated at 19 (normal range <5 pg/mL).
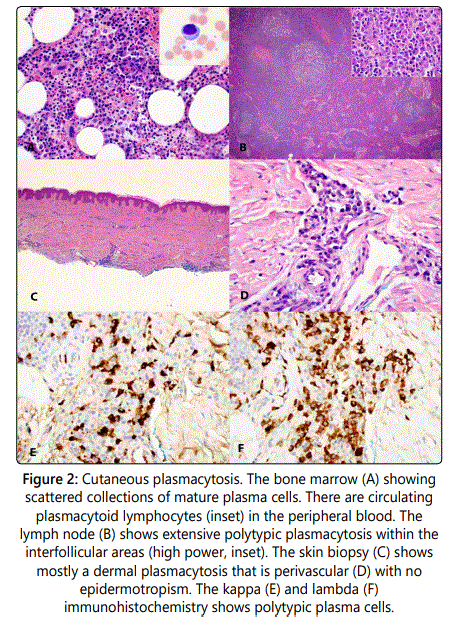
The patient presented to our clinic for a second opinion, where a broad shave biopsy of her most recent lesion, located on the right breast, was taken. This showed a perivascular, polytypic plasma cell infiltration without lymphomatous involvement or Castleman-like changes. Immunohistochemical stains were negative for HHV-8. Plasma cells displayed both kappa and lambda restriction, indicating polyclonality.
With the clinical presentation of multiple truncal red-brown plaques, the overall findings seen on skin biopsy were consistent with a diagnosis of cutaneous plasmacytosis. As the patient had self-discontinued her thalidomide due to severe fatigue, a new treatment strategy was pursued using rituximab infusions (375 mg/m2 every week for one month) and low dose prednisone.
The patient was treated with one month of rituximab at 375 mg/m2 (received 3 doses total) but did not showcase improvement. She was then attempted on tocilizumab (8 mg/kg) and received four monthly infusions. This resulted in improvement in the morphology of the patient's rash, particularly on the face and scalp, along with decreases in her ESR, CRP, and IL-6 levels. Rash on her back continues to persist. A repeat shave biopsy of a lesion on the patient's right upper back continued to show a perivascular, polytypic plasma cell infiltrate consistent with cutaneous plasmacytosis. The patient continues to receive follow up care to ensure no development of constitutional symptoms, or worsening of rash.
Comment
The condition now known as cutaneous and systemic plasmacytosis was first described in 1976 by Yashiro and colleagues [7-8] as a "kind of plasmacytosis." In 1980, Kitamura et al. [9] further refined the disease using the term "cutaneous plasmacytosis" [10]. Since then, it has been renamed as "cutaneous and systemic plasmacytosis (C/SP)" to reflect the high frequency of lymphoplasmacytic infiltration of extracutaneous sites [7]. In a minority of cases, C/SP is isolated to the skin alone. However, superficial lymphadenopathy often occurs in conjunction with skin findings in 58% of patients [7]. Other extracutaneous sites of plasma cell infiltration may include the liver, spleen, kidney, and the lung. A case of lymphoid interstitial pneumonia and a case of proliferative glomerulonephritis have been reported in the literature [1,7].
The extreme rarity of cutaneous and systemic plasmacytosis explains a lack of population-based studies; most information in the literature has been based primarily on case reports. The vast majority of C/SP has occurred in individuals of Japanese decent. However, cases have been reported in Chinese, Filipino, Korean, Hispanic, and Caucasian individuals [7]. A male to female ratio of 1:0.6, and a median age of presentation of 37 years has been reported [7,11].
Cutaneous manifestations of the disease appear to be similar across all cases. Characteristically, patients present in middle age with reddish-brown to purplish infiltrated macules, plaques, and flat tumors located on the trunk [1]. The appearance of the rash is often insidious in nature. Other accompanying symptoms may include fever, lymphadenopathy, and hepatosplenomegally [1].
Serum gammaglobulins are often elevated, along with IL-6, a cytokine responsible for inducing terminal differentiation of B cells into plasma cells [7]. Additionally, a normocytic anemia may be found.
Histopathology often reveals a dense, perivascular and perineural, mixed cell infiltrate with a predominance of mature plasma cells [1,7]. Infiltration occurs solely in the dermis and spares the epidermis. Polyclonality of the infiltrating plasma cells can be demonstrated with coexisting kappa and gamma-chain positive cells on immunohistochemical staining [7,10]. Furthermore, plasma cells do not showcase any degree of cellular atypia. Histology can mimic that of an extranodal, cutaneous, low-grade marginal-zone B cell lymphoma [1,7].
In presentations where lymphadenopathy occurs, lymph node biopsy most closely resembles a reactive germinal center [1]. It is important to perform accurate immunophenotyping of the lymph node to distinguish cutaneous and systemic plasmacytosis from follicular lymphoma [1]. In cutaneous marginal B cell lymphoma, follicular colonization by either monoclonal or polyclonal plasma cells has been described [1]. It remains unclear whether there is a relationship or spectrum of disease between C/SP and lymphoma. There are reports of C/SP patients developing malignant lymphomas, such as T cell lymphoma or non-Hodgkin's lymphoma. However, the relationship between C/SP and lymphoma currently remains unclear [1]. Transformation to plasma cell myeloma has not been reported [7].
The differential diagnosis for cutaneous and systemic plasmacytosis includes any disease entities resulting in plasma cell proliferation. These include chronic infection, collagen vascular disease, multiple myeloma, and cutaneous B cell lymphoma [7].
Due to the characteristic elevation of IL-6 seen in cutaneous and systemic plasmacytosis, some clinicians hypothesize that the disease is a variant of multicentric Castleman's disease (MCD). IL-6 induces B-cell proliferation and differentiation into plasma cells, immunoglobulin secretion, and angiogenesis [10]. IL-6 overproduction has been considered a key driving factor in the pathogenesis of MCD [10]. Castleman's disease was first reported in 1956 by Benjamin Castleman, who described a lymphoproliferative disorder in patients with mediastinal lymphadenopathy [10]. Multicentric Castleman's disease (MCD) is associated with lymphadenopathy, hyperproteinemia, hypergammaglobulinemia, increased erythrocyte sedimentation rate, and systemic symptoms such as fever, weight loss, and anemia. The plasma cell variant of cutaneous MCD results in skin histology that is similar to that of cutaneous and systemic plasmacytosis [10]. Interestingly, IL-6 elevation is not seen in all cases of C/SP, as some case reports reveal patients with normal levels despite clinical disease [12]. In a case where IL-6 was elevated at the time of disease presentation, treatment success was associated with a subsequent decrease in IL-6 levels [13].
Compared with multicentric Castleman's disease, most patients diagnosed with C/SP have a relatively favorable clinical course and prognosis [10]. However, a few cases of C/SP have followed an aggressive course resulting in fatal outcome [7]. Causes of death reported in the literature include leukemia, lymphoid interstitial pneumonia, and renal failure [7]. A few malignancies arising in patients with C/SP include T-cell lymphoma, and solid tumors such as lung, anal, and gastric cancer [10]. Uhara and colleagues suggest that a serum immunoglobulin level greater than 5000 mg/dL and plasma cell counts greater than 6.9% in the bone marrow are associated with more severe disease [14]. Though there are no consensus statements on follow-up, patients with a diagnosis of cutaneous or systemic plasmacytosis should be monitored longitudinally to assess for the possibility of malignant transformation [7].
The cause of cutaneous and systemic plasmacytosis is not known. A widespread hypothesis states that C/SP is a variant of reactive plasmacytic disorders [7]. Most cases have no detectible inciting cause. One case of C/SP noted the presence of skin lesions in a prior area of leg trauma [2]. The geographic distribution of C/SP cases with the majority occurring in Japan lead to the speculation that a primary infectious or environmental cause is responsible [7].
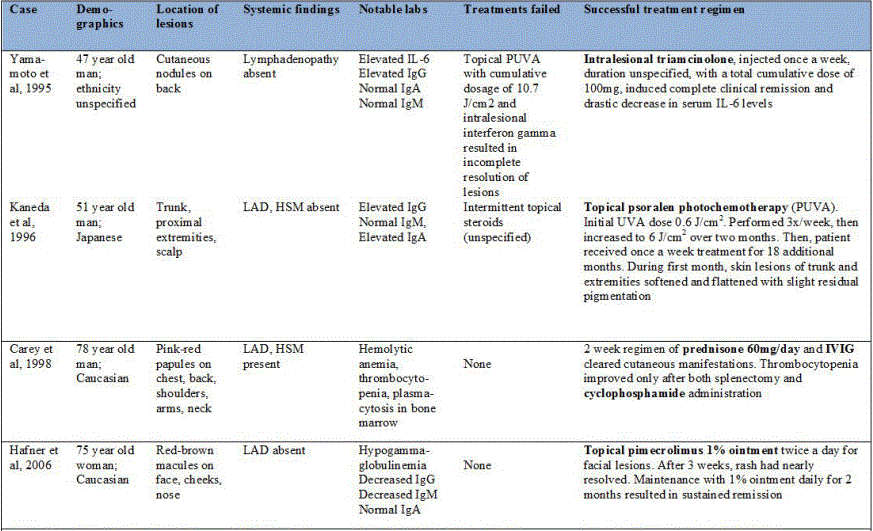
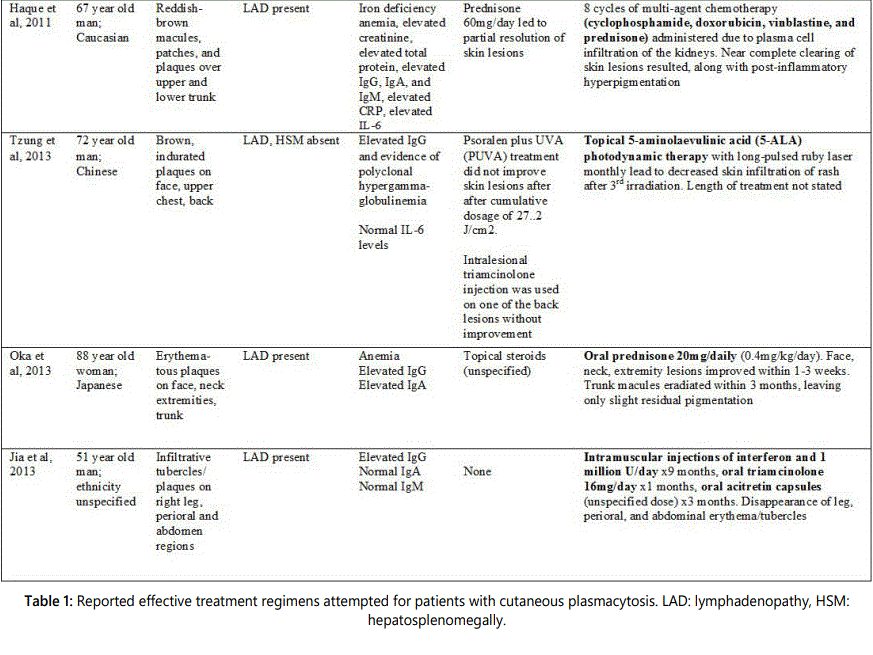
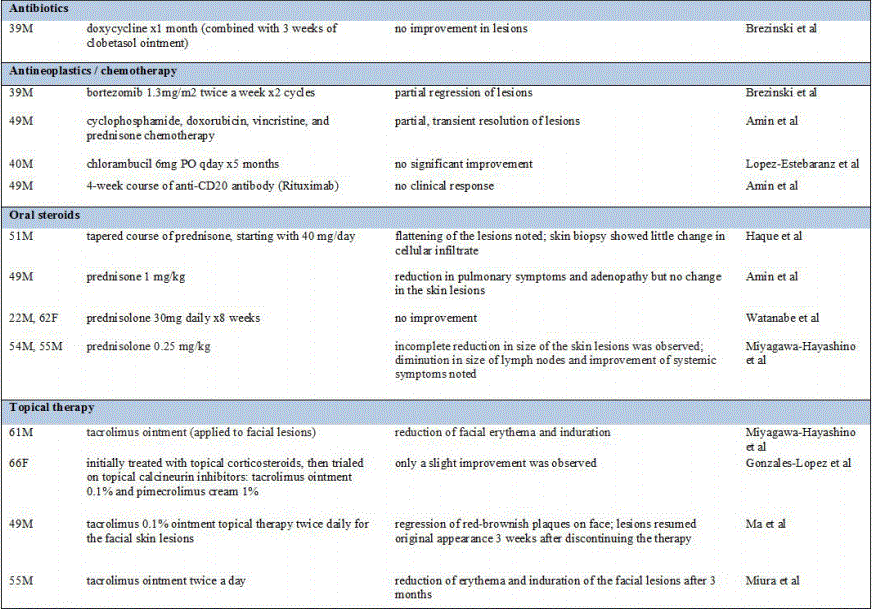
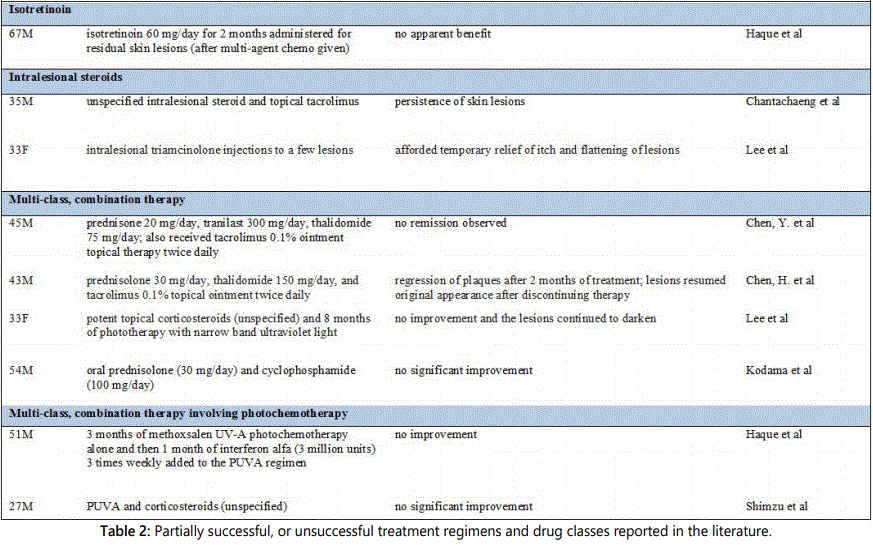
There is no uniform, conclusive method for the treatment of cutaneous and systemic plasmacytosis [2]. Topical and systemic steroids, radiation, topical tacrolimus, pimecrolimus, antibiotics, systemic chemotherapy, and anti-CD20 antibody therapy have been tried in C/SP with inconsistent results [10,11,14,15]. Only a handful of case reports describe significant improvement or remission in the treatment of C/SP. Table 1 represents cases published in the literature that have demonstrated notable treatment success.
Though rituximab, either alone or combined with cytotoxic agents, can induce sustained remissions in patients with multicentric Castleman's disease [16-18], there has been little experience with this drug in the treatment of cutaneous and systemic plasmacytosis. The interleukin-6 receptor (IL-6R) has been a viable target in multicentric Castleman's disease, where antagonism by the monoclonal antibody siltuximab, has shown favorable responses in a phase 2 study [19,20]. The safety and efficacy of tocilizumab was demonstrated in 28 symptomatic, HIV-negative patients with plasma cell type multicentric Castleman's disease. Patients were treated with tocilizumab 8mg/kg every two weeks for 16 weeks and noted that the drug alleviated lymphadenopathy and inflammatory markers. An additional subset of patients who had received oral corticosteroids prior to study entry were able to have their corticosteroid dose reduced without disease flare [21].
As both multicentric Castleman's disease and C/SP are associated with increased serum levels of IL-6, IL-6 antagonists may be an untapped resource for potential treatments for cutaneous and systemic plasmacytosis. Bortezomib, a 26S proteasome inhibitor used in the treatment of multiple myeloma and mantle cell lymphoma, is known to decrease the production of IL-6 via NFκB [19,22]. Brezinski and colleagues attempted treatment of antibiotic-refractory cutaneous plasmacytosis with twice weekly bortezomib. After two cycles, no new lesions developed, and persistent plaques had evidence of regression [3]. Ahmad et al describe a case of cutaneous Castleman's disease treated with a chimeric murine anti-human IL-6 antibody [23].
Pediatric Cases
A total of seven cases of cutaneous plasmacytosis in children have been described in the literature thus far. Interestingly, every case involved isolated skin lesions without the characteristic systemic symptoms seen in adults. Each pediatric case revealed uniform skin biopsy results, consisting of a dense, polyclonal plasma cell infiltrate in the superficial and mid dermis. A substantial fraction of pediatric cases involved lesions located on the extremities; this differs from the truncal distribution of lesions commonly seen in adults. The lack of systemic symptoms accompanying the skin lesions has prompted some authors to suggest that cutaneous plasmacytosis in children may be a completely separate entity from plasmacytosis in adults [24]. Others suggest that pediatric cutaneous plasmacytosis may be an example of a benign, cutaneous infiltrate of immune cells akin to entities such as incontinentia pigmenti, erythema toxicum neonatorum, Ofuji syndrome and eosinophilic pustular folliculitis [25,26].
Conclusion
Less than one hundred cases of cutaneous and systemic plasmacytosis have been documented in the literature. The lack of formal guidelines regarding treatment of this exceedingly rare disease necessitates a continued analysis of attempted treatments, whether successful or unsuccessful, in order to uncover possible patterns of efficacy. To our knowledge, the use of tocilizumab in the treatment of cutaneous plasmacytosis has not been documented, however its efficacy in multicentric Castleman's disease, a condition with many similarities to C/SP, is well known. Our patient did showcase symptomatic improvement after tocilizumab, with improved appearance of her rash, and notable decrease in her inflammatory markers. Interestingly, such improvements were not noted after three doses of rituximab. Due to the rarity of this disease, no randomized control trials exist comparing the efficacy of tocilizumab versus rituximab in the treatment of cutaneous plasmacytosis.
Our observations suggest that tocilizumab be considered as a potential efficacious regimen in the treatment of cutaneous plasmacytosis associated with elevated IL-6 levels.
References
Contact us for any additional information - contact@madridge.org
 Madridge Publishers is licensed under a Creative Commons Attribution 4.0 International License.
Madridge Publishers is licensed under a Creative Commons Attribution 4.0 International License.