

Review Article
Immune Checkpoint Inhibitors: A Step forward in Cancer Management
1Department of Biomedical Sciences and Human Oncology, University of Bari Aldo Moro, Policlinico, Piazza Giulio Cesare 11, Bari, Italy
2Institute of Biomembranes, Bioenergetics and Molecular Biotechnology, National Research Council (CNR), Bari, Italy
3Department of Basic Medical Sciences, Neurosciences and Sense Organs, University of Bari Aldo Moro, Policlinico, Piazza Giulio Cesare 11, Bari, Italy
*Corresponding author: Maria Rosaria Carratù, Department of Biomedical Sciences and Human Oncology, University of Bari Aldo Moro, Policlinico, Piazza Giulio Cesare 11, Bari, Italy, Tel: +39 080 5478455, E-mail: mariarosaria.carratu@uniba.it
Received: August 8, 2019 Accepted: September 4, 2019 Published: September 16, 2019
Citation: Carratù MR, Potenza MA, Nacci C, De Rasmo D, Signorile A. Immune Checkpoint Inhibitors: a Step Forward in Cancer Management. Madridge J Oncogenesis. 2019; 3(1): 65-72. doi: 10.18689/mjo-1000112
Copyright: © 2019 The Author(s). This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Cancer cells express unusual antigens which activate signal transduction pathways resulting in unregulated tumor growth. Hence, an effective immune response can be elicited via antibodies targeting the tumoral neoantigens. In particular, modulation of immunological pathways involved in the adaptive immune response is currently considered an innovative approach to fight against aggressive malignancies. Antigenspecific immune responses require critical interaction between antigen presenting cells (APC), T-lymphocytes, and target cells. Upon T-cell activation, the programmed cell death protein-1 (PD-1) blocks the early TCR/CD28 signalling pathway, thus hindering cytokine production and cell cycle progression. Furthermore, cytotoxic T-lymphocyte antigen-4 (CTLA-4) inhibits the activated T-cell by binding with high affinity to the B7 molecules and hampering the CD28-mediated signalling. The characterization of these negative immune regulators led to the successful development of fully human monoclonal antibodies (mAbs), specifically designed to promote T-cell activation and increase the anti-tumor immune response. This article provides a brief overview of immune modulation of T-cell function by anti-CTLA-4 and anti-PD-1 antibodies, known as checkpoint inhibitors, and summarizes clinical benefits and limitations of their use in cancer management.
Keywords: T-cell; CTLA-4; PD-1; Immunotherapy; Cancer; Immune checkpoint inhibitors.
Introduction
Cancer cells express unusual neoantigens which activate signal transduction pathways resulting in unregulated tumor growth. The paradigm is represented by erythroblastic oncogene B (ErbB2), a constitutively active cell surface receptor over-expressed on the surface of breast cancer cells [1]. Indeed, there are several tumor-associated antigens targeted by mAbs. They include cell surface differentiation antigens expressed in normal and in tumor cells, growth factors and their receptors, stromal and extracellular matrix antigens [2].
Once bound to their antigens, mAbs interact in several ways with the immune systems through their Fc portion, thereby triggering complement-dependent or antibody-dependent cytotoxicity, and phagocytosis of target cells. Additionally, more specific mAbs have been tailored to modulate immunological pathways involved in the immune surveillance, and this strategy is considered an innovative approach to fight against aggressive malignancies [3]. The immune system capacity to recognize and kill tumor cells is strongly supported by evidence accumulated over the last ten years from preclinical and clinical studies. It is currently accepted that malignancy develops and progresses as a result of the adaptive immune response circumvention [4]. Hence, the immune system failure in the control of tumor progression determines the selection of variants of tumor cells that, in turn, repress the antitumor immune response [5]. The phases of cancer immunoediting, that is the capability of the immune system to suppress or promote tumor growth, have been extensively reviewed [6]. Negative regulatory pathways, known as immune checkpoints, have a critical function in both peripheral and central tolerance. They may act at each stage of the immune response. Under persistent antigenic burden T-cells upregulate the immune checkpoints, whose signalling blocks their proliferation and effector functions, eventually leading to their deletion [7]. There is compelling evidence that the immune exhaustion also occurs in cancer [8,9]. Several T-cell surface receptors with inhibitory activity have been identified, and some of them, such as CTLA-4 and PD-1, are targets of mAbs already approved for cancer therapy.
The Immune Checkpoint receptors CTLA-4 and PD-1 modulate the T-Cell Effector functions
Antigen-specific immune responses require antigen presenting cells (APC), T-lymphocytes and target cells interaction. The recognition of specific antigens bound to major histocompatibility complex (MHC) by T-cell receptor (TCR) is not sufficient per se to activate T-cells. T-cell activation requires additional co-stimulation through binding of CD28 with molecules of the B7 family (CD80, CD86), thus triggering signalling pathways eliciting autocrine IL-2 production.
Co-stimulatory signal is inhibited by CTLA-4 through competition with CD28
The inhibition of T-cell activation by CTLA-4 occurs by two mechanisms and depends on its expression on cell surface:delivery of a negative signal and competitive antagonism of CD28:B7-mediated co-stimulation [10]. So far, the molecular mechanism that controls negative signalling is unknown. There is evidence that negative signalling needs the CTLA-4 cytosolic tail and, further, a low expression at the cell surface, thus leading to a rapid inhibition of T-cell activation [11]. The other mechanism proposes that, subsequently to the activation of T-cells, CTLA-4 moves from intracellular compartments to the immunological synapse thus inhibiting the costimulatory signal by competing with CD28 for binding to CD80 on the APC [12,13]. CTLA-4 has a markedly higher affinity for CD80 (B7) compared with CD28 (Kd 12 nM versus 200 nM) [14]. This higher binding affinity depends on a high degree of shape complementarity resulting in unusually stable CTLA-4/CD80 signalling complexes at the T-cell surface [15]. Constitutive expression of CD28 has been observed in resting and activated cells, while the expression of CTLA-4 is regulated by T-cell receptor (TCR)/CD28 co-stimulation. Levels of CD28 expression on naïve and active T-cells are influenced by the rate of biosynthesis and mechanisms that control its internalization. There is evidence that phosphatidylinositol 3-kinase (PI3K) is the main regulatory factor of CD28 endocytosis that occurs in the form of CD28–PI3K complexes [16-18]. CTLA-4 is localized in Trans Golgi network (TGN), lysosome, and endosomes, [19-21], and a small amount has been detected on cell surface even after the activation of T-cells. The mechanism involved in the cell surface translocation should not be underestimated, given that even minimal expression differences can have remarkable consequences on the development of autoimmunity and the function of T-cells. Despite these findings, components of the complex array controlling the surface transport have not been entirely identified so far. Indeed, a type III transmembrane protein of 30 kDa, called TCR interacting molecule (TRIM), seems to regulate this event in a chaperone manner, since overexpression of TRIM has been found closely related to the increased CTLA-4 translocation at the cell surface [21]. It is evident that the CD28/CTLA-4 axis represents a viable way to modulate the immune system response. Abrogation of the CTLA-4 function by means of mAbs would restore CD28 signalling and switch the balance in favor of immune stimulation. The characterization of this negative immune regulator led to the successful development of ipilimumab and tremelimumab, two fully human monoclonal antibodies, designed to potentiate T-cell activation and increase the antitumor immune response. Only ipilimumab was approved for metastatic melanoma treatment.
PD-1 negatively regulates T-cell receptor signals by inducing CD28 dephosphorylation
The programmed cell death protein 1 (PD-1), which is homologous to CD28, is a negative regulator of adaptive immune responses [22]. PD-1 is a receptor at cell surface, belonging to the superfamily of immunoglobulin that is expressed in T-cells and, unlike CTLA-4, also in activated B cells and macrophages [23]. This suggests a wider function of PD-1, compared to CTLA-4, in the immune response regulation. Structurally, PD-1 presents an extracellular IgVlike ligand-binding domain, a transmembrane region and a cytoplasmic tail containing two phosphorylation sites located in an immunoreceptor tyrosine-based inhibitory motif and an immunoreceptor tyrosine-based switch motif [24,25], which recruit SHP-1 and SHP-2 phosphatases upon ligand binding. Unlike some T-cell populations such as follicular helper T-cells, in which PD-1 is constitutively expressed [26], in the most circulating T-cells PD-1 is expressed upon stimulation, through the TCR complex or exposure to growth factor (TGF)-β and cytokines [23,27]. The two ligands of the receptor, PD-L1 (B7-H1; CD274) and PD-L2 (B7-DC; CD273), can be detected on the APC surface, and are up-regulated by interferon (INF) γ [28]. In T-cells, activation of PD-1 triggers a signalling cascade leading to the dephosphorylation of TCR signalling components [29]. CD28 seems to be the primary target [30] and plays, upon the TCR stimulation, a main role in the activation of T-cells. By interfering with TCR/CD28 pathways, PD-1 signalling cascade reduces cytokine production and inhibits IL-2-dependent cell cycle progression. Thus, PD-1 activity is relevant only during T-cell activation and is crucial for restraining immune responses so as to avoid immunopathology. Importantly, genetic polymorphisms in the PD-1 locus have been associated with a higher susceptibility towards autoimmune diseases [31,32].
PD-1 is up-regulated in circulating tumor-specific T-cells and tumor-infiltrating lymphocytes, where it is associated with decreased T-cell functions [33-35]. Moreover, PD-1-positive dendritic cells, with a decreased capacity to induce T-cell stimulation, have been identified in hepatocellular carcinoma [36], and tumor infiltration of PD-1 positive regulatory B cells producing IL-10 is associated with poor prognosis [37]. Importantly, both murine and human tumorassociated macrophages (TAM) can also express high levels of PD-1 with consequent impairment in phagocytosis and tumor immunity, whereas the deletion of PD-L1 enhances PD-1+ TAM phagocytosis and reduces tumor growth [38]. Hence, the effects of PD-1 blockade with anti-PD-1/PD-L1 antibodies on macrophages in human cancer may lead to novel therapeutic strategies.
The Repertoire of Checkpoint Inhibitors currently Available
The improved progress-free survival and overall survival of patients have highlighted the clinical benefit of both CTLA4 and PD-1 checkpoint inhibitors. Unlike traditional cytotoxic therapies, which directly attack and kill tumor cells, immunotherapies promote the anti-tumor immune response. Tumor cells contrive several mechanisms to escape immuneme-diated destruction. The updated concept of cancer immunoediting well explains the involvement of immune system in the development or regression of tumor. Conceptually, cancer immunoediting, extensively reviewed by Teng et al. [6], may be outlined into elimination, equilibrium, and escape phases. In particular, multiple mechanisms may be responsible for tumor cell escape including (i) loss of tumor antigens and/or derangement of the T-cell activation machinery; (ii) over-expression of prosurvival or growth factors (e.g., Bcl-2, Her2/neu); (iii) setting up of an immunosuppressive tumor microenvironment (TME) through production of cytokines and metabolic factors, or through recruitment of regulatory T-cells (TREG), and involvement of negative regulatory pathways, such as CTLA-4 and PD-1. In recent years, strategies to target inhibitory receptors have been extensively investigated, and anti-CTLA-4 and anti-PD-1 antibodies gained regulatory approval for human use due to remarkable improvement observed in disease outcomes, especially in melanoma patients.
Anti CTLA-4 therapeutics: the first step forward in melanoma treatment
Melanoma, a skin cancer originating in melanocytes, is associated with a high mortality rate [39]. Currently, for patients with melanoma, intolerant to first-line treatments or affected by progressive disease [40,41], there is no standard therapy. Temozolomide, an analogue of dacarbazine, is often used off-label in the treatment of metastatic melanoma taking advantage of its oral availability while retaining the same efficacy as dacarbazine [42,43]. Tumor-specific neoantigens are often expressed in melanoma cells, thus remarkable therapeutic effects may be attained using immunotherapeutic agents which activate cytotoxic T lymphocytes [41]. Indeed, tremelimumab and ipilimumab, two monoclonal antibodies, have been investigated for the advanced melanoma treatment.
Ipilimumab is a fully human monoclonal IgG1 antibody that targets CTLA-4 preventing its interaction with CD80 and CD86 (B7 molecules). CTLA-4, by down-regulating T-cell activation pathways and preventing autoimmunity, plays the role of “immune checkpoint” [12]. Ipilimumab removes the CTLA-4-mediated negative immune regulation resulting in unrestrained T-cell proliferation [3,44-47]. Thus, the beneficial effect of ipilimumab in melanoma patients lies in T-cell mediated antitumor immune responses (Figure 1). Phase III studies in metastatic melanoma showed that ipilimumab improves overall survival of patients with disease progression on prior therapy [47]. This therapeutic success led to the FDA approval of ipilimumab for the treatment of metastatic melanoma in 2011, thus marking the beginning of new era in cancer immunotherapy [39]. Conversely, phase III studies of tremelimumab, an IgG2 isotype developed to mitigate complement activation and the risk of cytokine release syndrome [48], were discontinued in 2008, since patients with advanced melanoma did not benefit from the tremelimumab treatment [39].
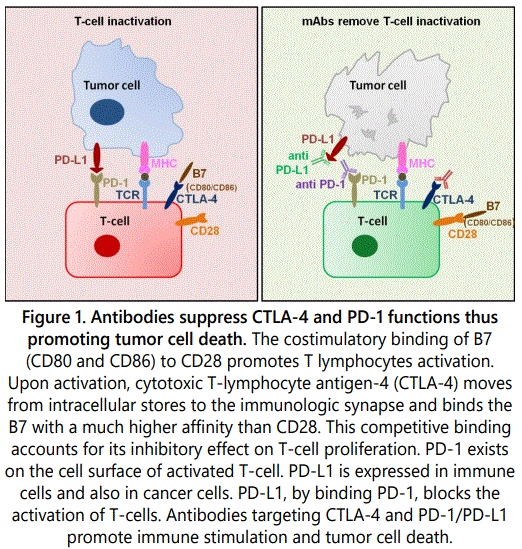
Anti PD-1/PD-L1 therapeutics: their benefit extends from melanoma to other types of tumors
PD-L1, the ligand for PD-1, is highly expressed in several cancers, thus the PDL1/PD-1 interaction activates signalling pathways that play a fundamental role in cancer immune evasion [49-51]. Prevention of the PDL1/PD-1 interaction by means of mAbs elicits T-cell responses and restores the immune-mediated antitumor activity (Figure 1). Some anticancer mAbs targeting the PD-1 receptor have been already approved. Details of the molecular mechanism involved in tumor responsiveness to anti-PD1 treatment are still unknown. Although cancer cells expressing PD-L1 are more responsive to the treatment [52,53], PD-L1 negative tumors may also partially respond to anti-PD-1 treatment, suggesting that PD-L1 expression in the TME is not the unique factor associated with clinical response to anti-PD-1 therapy [53].
Nivolumab, a fully human IgG4 anti-PD-1 antibody, produced a high response rate in multiple tumor types, including non-small-cell lung cancer (NSCLC), metastatic melanoma, and renal-cell cancer, as shown by results from a phase I clinical trial enrolling 296 patients [54]. In 2014, nivolumab was approved for metastatic melanoma.
An emerging and promising approach to fight against aggressive malignancies is based on the combination of anti PD-1 and anti CTLA-4 antibodies with additive benefit [55-57]. Anti-CTLA-4 treatment enhances antigen specific T-cell-dependent immune reaction on one side and on the other, anti-PD-1 treatment reactivates CD8+ T-cells thus promoting the death of cancer cells [49,58,59]. In fact, clinical studies have shown that patients refractory to single blockade are more responsive to combination therapy [52]. A treatment regimen combining anti-CTLA-4 (ipilimumab) with anti-PD-1 (nivolumab) gained FDA approval for melanoma in 2015 [53]. Pembrolizumab, a humanized monoclonal IgG4 antibody against PD-1, was approved by the FDA in September 2014 for metastatic melanoma, and in October 2015 for advanced (metastatic) NSCLC patients with disease progression on prior therapy [57]. Additional drugs targeting PD-1 receptors, such as pidilizumab, BMS-936559, and avelumab, is still under evaluation.
Augmented levels of PD-L1 have been associated with poor prognosis in melanoma and also in other malignancies such as ovarian, lung, renal and nasopharyngeal cancers [60-64]. Inhibition of PD-L1 with antibodies improved overall survival in patients with these cancers. More recently, the mAb against PD-L1, atezolizumab, has been approved for the treatment of PD-L1 positive bladder cancer and NSCLC [65,66]. The PD-L1 expression is under the control of MAPK and PI3K/Akt signaling pathways and transcriptional factors HIF-1 and STAT3 [67]. Therefore, the inhibition of signaling pathways, that promote cancer development by targeting a number of downstream effectors, could also downregulate PD-L1 expression. Indeed, there is evidence that the use of the signaling inhibitor trametinib to reduce PD-L1 expression together with anti-PD-1 antibodies results in a greater efficacy [68].
Safety Profile and Limitations of Immune Checkpoint inhibitors
Treatment with immune checkpoint inhibitors results in adverse events which are closely related to their mechanisms of action. PD-1 and CTLA-4 are key regulators of the adaptive immune response. Hence, inhibition of these regulatory pathways through anti PD-1 and anti CTLA-4 antibodies for the cancer treatment is associated with a high and unavoidable risk of immune-related adverse events (irAEs). Of note, inflammation plays a critical role in tumorigenesis and can contribute to tumor promotion. Hence, cancer immunotherapy that induces pro-inflammatory cytokines may be expected to contribute to tumorigenesis [69].
PD-1 and CTLA-4 inhibition is associated with a substantial risk of irAEs
Clinical trials have shown that more than 80% of patients receiving ipilimumab develop adverse events [47,70-74], with a high rate (10% to 26%) of grade 3-4 drug-related unwanted reactions [71,73,74]. Hence, the high risk of ipilimumab use in patients with severe autoimmune disorders [39]. Although any organ system may be affected, the most common severe irAEs are dermatitis (including potentially fatal epidermal necrolysis), hepatitis, enterocolitis, endocrinopathy and neuropathy [75]. The range of side effects may expand to rarer manifestations involving nervous, hematopoietic and urinary systems [76]. However, most of the ipilimumab immune-mediated side effects are reversible if diagnosed and treated early [77]. Clinical trials comparing different inhibitors of immune checkpoints revealed that the incidence of side effects is higher in patients treated with anti-CTLA-4 (27.3%) with respect to those treated with anti-PD-1 (16.3%) [78]. Moreover, the percentage increases to 55% if a combination of antibodies is used [78]. The reduced incidence of adverse effects observed in patients treated with anti-PD-1 antibodies might be explained taking into account that PD-1 suppresses T-cell activity mainly in the peripheral tissues and TME, whereas CTLA-4 modulates the activation of T-cells in the lymphoid organs where naïve T-cells are primed, and potentially in the periphery via TREG depletion [75]. The diverse pattern of CTLA-4 action in modulating immune responses might explain the wide range of irAEs observed in patients treated with anti-CTLA-4 antibodies.
Why the treatments with anti-PD-1 and anti-CTLA-4 lose efficacy
One important limitation of treatments with immune checkpoint inhibitors lies on the loss of efficacy after a period of time, thus many patients with initial responses frequently relapse. Several studies have analyzed the mechanisms underlying the loss of efficacy. Tumor cells can evade from immune surveillance through vicarious inhibitory pathways up-regulated by anti-PD-1 or anti-CTLA-4 therapeutic antibodies [7]. Concerning anti-CTLA-4 treatment, up-regulation of the inhibitory receptor V-domain Ig suppressor of T-cell activation (VISTA) has been observed on tumor-infiltrating immune cells in patients affected by melanoma or prostate cancer [79]. Moreover, in adenocarcinoma patientʼs refractory to anti-PD-1 treatment, up-regulation of the inhibitory receptor TIM-3 (T-cell immunoglobulin and mucin-domain containing-3) has been detected on the T-cell surface [80]. Yet, acquired resistance to anti-PD-1 treatment in melanoma patients has been ascribed to the altered INF-γ receptor signaling pathway and antigen presentation [80]. A better knowledge of the molecular mechanisms involved in the efficacy loss of immune checkpoint inhibitors will significantly contribute to improve the therapeutic approach in the near future.
The need for biomarkers to avoid inappropriate drug exposure
A significant number of patients do not benefit from antiPD-1 and anti-CTL-A4 therapy, hence predictive biomarkers are needed to assess responsiveness to immune checkpoint inhibitors. Basically, factors with predictive positive value include small size tumor or low serum lactate dehydrogenase (LDH) [81-83]. Patients with reduced LDH levels are more responsive to anti-PD-1 treatment [84]. Dead tumor cells release circulating tumor DNA (ctDNA) containing melanoma-associated mutations. High ctDNA serum levels have negative predictive value and correlate with tumor progression [85,86]. The expression of neoantigens on mutated tumor cells improves efficacy of anti-CTLA-4 treatment in melanoma patients [56].
Immunological biomarkers in the context of anti-PD1 therapy for melanoma can be represented by CD8+ T-cell density at tumor edge that has been correlated to clinical response to anti-PD1 treatment [87]. In addition, PD-L1 expression on tumor cells correlates with efficacy of anti-PD1 therapy in melanoma patients [54,78], as well as patients with several types of solid tumors [88]. However, in multiple tumor biopsies collected from different anatomical sites in individual patients, variable expression of PD-L1 could be observed. This rises a pitfall in considering PD-L1 a biomarker when it is measured by immunohistochemistry (IHC) in a single tumor specimen per patient [54]. In any case, in October 2015, FDA approved PD-L1 IHC test as diagnostic test to follow the treatment-response of patients with NSCLC. Genetic markers associated with resistance to checkpoint blockade and relapse have been extensively reviewed [89]. A correlation between DNA mismatch repair complex genotype and response to anti-PD-1 therapy is reported [90]. The anti-PD-1 treatment is more effective in patients with colorectal cancer or NSCLC with mismatch repair deficiency or high mutation rates [90,91].
In patients resistant to PD-1 inhibition, Hugo and coworkers [92] uncovered, in distinctive histological subtypes of advanced cancer, an ‘innate anti-PD-1 resistance’ characterized by an increased expression of genes encoding for factors involved in monocyte/macrophage chemotaxis, epithelial mesenchymal transition, extracellular matrix remodelling, wound healing, cellular adhesion, and angiogenesis. The key point emerging from all studies is the critical role of tumor gene-expression profiling for the most appropriate choice of therapeutic strategies matched to patients and tumor characteristics, and also for prediction of immunotherapeutic response.
Unlike PD-1/PD-L1, the CTLA-4 immune checkpoint predominantly acts in the early steps of the immune response cascade, during T-cell priming and activation, and it enhances the immunosuppressive activity of TREG cells. Hence, CTLA-4 exhibits a general impact on the immune system, and consequently biomarkers of resistance/response to anti-CTLA-4, different from those of anti-PD1-PDL1 drugs, should be identified.
Conclusions
Immunotherapy with anti-PD-1 or anti-CTLA-4 mABs has drastically improved the management of patients with advanced-stage melanoma and is also a promising strategy for many other types of cancer. The anti-CTLA-4 antibody ipilimumab was the first checkpoint inhibitor with ascertained survival benefit in advanced melanoma, and the first gaining FDA approval. The initial enthusiasm for the discovery and delivery of this novel drug was mitigated by concerns about the unavoidable risk of severe immune-mediated adverse effects [93]. Despite that, ipilimumab marked the beginning of a new era of melanoma therapy after the repeated failure of traditional cytotoxic agents. Subsequently, mABs targeting PD-1 were developed and approved for the treatment of melanoma and other types of cancer. Actually, the mortality rate remains high among patients with aggressive malignancy, thus underlying the need for development of additional innovative drugs. To enhance anti-tumor immune responses and to select therapeutic approaches matched to patients and tumor characteristics, genomic-driven precision immunotherapy will likely be a winner strategy. Although significant response rates have been reported with checkpoint blockade in melanoma, NSCLC, bladder cancer and Hodgkinʼs lymphoma [94], the clinical benefit of these inhibitors is still restricted to a subset of patients and certain types of tumors [53,72,95]. In this scenario, it is mandatory to validate specific biomarkers in order to assess therapeutic response, and also to minimize adverse events especially for drugs, such as ipilimumab, with an unfavourable risk-benefit balance. Concerning anti-PD-1 therapy, biomarkers are incredibly useful for ranking exceptionally responsive patients. For example, advanced colorectal tumors generally do not respond to anti-PD-1 therapy, but a subset with genomic instability and high tumor mutational burden is very responsive [90]. Finally, we should also consider that the number of checkpoint receptors and ligands is increasing. Some of these are co-expressed with PD-L1, hence providing a rationale for dual block therapy. Validated biomarkers to predict the benefits of the dual block therapy are not available so far. Despite explosion of data in the immunotherapy field, selection of patients for mono- or combination therapy, and guide clinical decision-making for treatment sequencing still pose enormous challenges to researchers and require further efforts to implement preclinical and clinical approaches.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Omenn GS, Guan Y, Menon R. A new class of protein cancer biomarker candidates: differentially expressed splice variants of ERBB2 (HER2/neu) and ERBB1 (EGFR) in breast cancer cell lines. J Proteomics. 2014; 107: 103-112. doi: 10.1016/j.jprot.2014.04.012
- Scott AM, Allison JP, Wolchok JD. Monoclonal antibodies in cancer therapy. Cancer Immun. 2012; 12: 14.
- Hwu P. Treating cancer by targeting the immune system. N Engl J Med. 2010; 363(8): 779-781. doi: 10.1056/NEJMe100641
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144(5): 646-674. doi: 10.1016/j.cell.2011.02.0136
- Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunityʼs roles in cancer suppression and promotion. Science. 2011; 331(6024): 1565-1570. doi: 10.1126/science.1203486
- Teng MW, Galon J, Fridman WH, Smyth MJ. From mice to humans: developments in cancer immunoediting. J Clin Invest. 2015; 125(9): 3338-3346. doi: 10.1172/JCI80004
- Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front Oncol. 2018; 8: 86. doi: 10.3389/fonc.2018.00086
- Day CL, Kaufmann DE, Kiepiela P, et al. PD-1 expression on HIV-specific T-cells is associated with T-cell exhaustion and disease progression. Nature. 2006; 443(7109): 350-354. doi: 10.1038/nature05115
- Baitsch L, Baumgaertner P, Devêvre E, et al. Exhaustion of tumor-specific CD8⁺ T-cells in metastases from melanoma patients. J Clin Invest. 2011; 121(6): 2350-2360. doi: 10.1172/JCI46102.
- Carreno BM, Bennett F, Chau TA, et al. CTLA-4 (CD152) can inhibit T-cell activation by two different mechanisms depending on its level of cell surface expression. J Immunol. 2000; 165(3): 1352-1356. doi: 10.4049/jimmunol.165.3.1352
- Madrenas J, Chau LA, Teft WA, et al. Conversion of CTLA-4 from inhibitor to activator of T-cells with a bispecific tandem single-chain Fv ligand. J Immunol. 2004; 172(10): 5948-5956. doi: 10.4049/jimmunol.172.10.5948
- Melero I, Hervas-Stubbs S, Glennie M, Pardoll DM, Chen L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat Rev Cancer. 2007; 7(2): 95-106. doi: 10.1038/nrc2051
- Nomura T, Kabashima K, Miyachi Y. The panoply of αβT-cells in the skin. J Dermatol Sci. 2014; 76(1): 3-9. doi: 10.1016/j.jdermsci.2014.07.010
- van der Merwe PA, Bodian DL, Daenke S, Linsley P, Davis SJ. CD80 (B7-1) binds both CD28 and CTLA-4 with a low affinity and very fast kinetics. J Exp Med. 1997; 185(3): 393-403. doi: 10.1084/jem.185.3.393
- Stamper CC, Zhang Y, Tobin JF, et al. Crystal structure of the B7-1/CTLA-4 complex that inhibits human immune responses. Nature. 2001; 410(6828): 608-611. doi: 10.1038/35069118
- Prasad KV, Cai YC, Raab M, et al. T-cell antigen CD28 interacts with the lipid kinase phosphatidylinositol 3-kinase by a cytoplasmic Tyr(P)-MetXaa-Met motif. Proc Natl Acad Sci U S A. 1994; 91(7): 2834-2838. doi: 10.1073/pnas.91.7.2834
- Pagès F, Ragueneau M, Rottapel R, et al. Binding of phosphatidylinositol3-OH kinase to CD28 is required for T-cell signalling. Nature. 1994; 369(6478): 327-329. doi: 10.1038/369327a0
- August A, Dupont B. CD28 of T lymphocytes associates with phosphatidylinositol 3-kinase. Int Immunol. 1994; 6(5): 769-774. doi: 10.1093/intimm/6.5.769
- Linsley PS, Bradshaw J, Greene J, Peach R, Bennett KL, Mittler RS. Intracellular trafficking of CTLA-4 and focal localization towards sites of TCR engagement. Immunity. 1996; 4(6): 535-543.
- Alegre ML, Noel PJ, Eisfelder BJ, et al. Regulation of surface and intracellular expression of CTLA4 on mouse T-cells. J Immunol. 1996; 157(11): 4762-4770.
- Valk E, Leung R, Kang H, Kaneko K, Rudd CE, Schneider H. T-cell receptorinteracting molecule acts as a chaperone to modulate surface expression of the CTLA-4 coreceptor. Immunity. 2006; 25(5): 807-821. doi: 10.1016/j.immuni.2006.08.024
- Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupuslike autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999; 11(2): 141-151.
- Agata Y, Kawasaki A, Nishimura H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. 1996; 8(5): 765-772. doi: 10.1093/intimm/8.5.765
- Ishida Y, Otsuka A, Tanaka H, Levesque MP, Dummer R, Kabashima K. HLA-A*26 is Correlated with response to Nivolumab in Japanese Melanoma Patients. J Invest Dermatol. 2017; 137(11): 2443-2444. doi: 10.1016/j.jid.2017.06.023
- Blank C, Mackensen A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: an update on implications for chronic infections and tumor evasion. Cancer Immunol Immunother. 2007; 56(5): 739-745. doi: 10.1007/s00262-006-0272-1
- Sage PT, Francisco LM, Carman CV, Sharpe AH. The receptor PD-1 controls follicular regulatory T-cells in the lymph nodes and blood. Nat Immunol. 2013; 14(2): 152-161. doi: 10.1038/ni.2496
- Kinter AL, Godbout EJ, McNally JP. The common gamma-chain cytokines IL-2, IL-7, IL-15, and IL-21 induce the expression of programmed death-1 and its ligands. J Immunol. 2008; 181(10): 6738-6746. doi: 10.4049/jimmunol.181.10.6738
- Brown JA, Dorfman DM, Ma FR, et al. Blockade of programmed death-1 ligands on dendritic cells enhances T-cell activation and cytokine production. J Immunol. 2003; 170(3): 1257-1266. doi: 10.4049/jimmunol.170.3.1257
- Patsoukis N, Brown J, Petkova V, Liu F, Li L, Boussiotis VA. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T-cell proliferation. Sci Signal. 2012; 5(230): ra46. doi: 10.1126/scisignal.2002796
- Hui E, Cheung J, Zhu J, et al. T-cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017; 355(6332): 1428-1433. doi: 10.1126/science.aaf1292
- Nielsen C, Hansen D, Husby S, Jacobsen BB, Lillevang ST. Association of a putative regulatory polymorphism in the PD-1 gene with susceptibility to type 1 diabetes. Tissue Antigens. 2003; 62(6): 492-497. doi: 10.1046/j.1399-0039.2003.00136.x
- Velázquez-Cruz R, Orozco L, Espinosa-Rosales F, et al. Association of PDCD1 polymorphisms with childhood-onset systemic lupus erythematosus. Eur J Hum Genet. 2007; 15(3): 336-341. doi: 10.1038/sj.ejhg.5201767
- Ahmadzadeh M, Johnson LA, Heemskerk B, et al. Tumor antigen-specific CD8 T-cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009; 114(8): 1537-1544. doi: 10.1182/blood-2008
- Chapon M, Randriamampita C, Maubec E, et al. Progressive upregulation of PD-1 in primary and metastatic melanomas associated with blunted TCR signaling in infiltrating T lymphocytes. J Invest Dermatol. 2011; 131(6): 1300-1307. doi: 10.1038/jid.2011.30
- Saito H, Kuroda H, Matsunaga T, Osaki T, Ikeguchi M. Increased PD-1 expression on CD4+ and CD8+ T-cells is involved in immune evasion in gastric cancer. J Surg Oncol. 2013; 107(5): 517-522. doi: 10.1002/jso.23281
- Lim TS, Chew V, Sieow JL, et al. PD-1 expression on dendritic cells suppresses CD8+ T-cell function and antitumor immunity. Oncoimmunology. 2015; 5(3): e1085146. doi: 10.1080/2162402X.2015.1085146
- Xiao X, Lao XM, Chen MM, et al. PD-1hi identifies a Novel Regulatory B-cell Population in Human Hepatoma that promotes Disease Progression. Cancer Discov. 2016; 6(5): 546-559. doi: 10.1158/2159-8290.CD-15-1408.
- Gordon SR, Maute RL, Dulken BW, et al. PD-1 expression by tumourassociated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017; 545(7655): 495-499. doi: 10.1038/nature22396
- Fellner C. Ipilimumab (Yervoy) prolongs survival in advanced melanoma: serious side effects and a hefty price tag may limit its use. P T. 2012; 37(9): 503-530.
- Culver ME, Gatesman ML, Mancl EE, Lowe DK. Ipilimumab: A novel treatment for metastatic melanoma. Ann Pharmacother. 2011; 45(4): 510-519. doi: 10.1345/aph.1P651
- Sondak VK, Smalley KS, Kudchadkar R, Grippon S, Kirkpatrick P. Ipilimumab. Nat Rev Drug Discov. 2011; 10(6): 411-412. doi: 10.1038/nrd3463
- Quirbt I, Verma S, Petrella T, Bak K, Charette M. Temozolomide for the treatment of metastatic melanoma. Curr Oncol. 2007; 14(1): 27-33. doi: 10.3747/co.2007.98
- Fricker J. New era in metastatic melanoma. Mol Oncol. 2010; 4(2): 91-97. doi: 10.1016/j.molonc.2010.02.001
- OʼDay SJ, Hamid O, Urba WJ. Targeting cytotoxic T-lymphocyte antigen-4 (CTLA-4): A novel strategy for the treatment of melanoma and other malignancies. Cancer. 2007; 110(12): 2614-2627. doi: 10.1002/cncr.23086
- Fong L, Small EJ. Anti-cytotoxic T-lymphocyte antigen-4 antibody: The first in an emerging class of immunomodulatory antibodies for cancer treatment. J Clin Oncol. 2008; 26(32): 5275-5283. doi: 10.1200/JCO.2008.17.8954
- Robert C, Ghiringhelli F. What is the role of cytotoxic T lymphocyteassociated antigen 4 blockade in patients with metastatic melanoma? Oncologist. 2009; 14(8): 848-861. doi: 10.1634/theoncologist.2009-0028
- Hodi FS, OʼDay SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010; 363(8): 711-723. doi: 10.1056/NEJMoa1003466
- Ribas A, Hanson DC, Noe DA, et al. Tremelimumab (CP-675,206), a cytotoxic T lymphocyte associated antigen 4 blocking monoclonal antibody in clinical development for patients with cancer. Oncologist. 2007; 12(7): 873-883. doi: 10.1634/theoncologist.12-7-873
- Syn NL, Teng MWL, Mok TSK, Soo RA. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017; 18(12): e731-e741. doi: 10.1016/S1470-2045(17)30607-1
- Wang X, Teng F, Kong L, Yu J. PD-L1 expression in human cancers and its association with clinical outcomes. Onco Targets Ther. 2016; 9: 5023-5039. doi: 10.2147/OTT.S105862
- Gandini S, Massi D, Mandalà M. PD-L1 expression in cancer patients receiving anti PD-1/PD-L1 antibodies: A systematic review and meta-analysis. Crit Rev Oncol Hematol. 2016; 100: 88-98. doi: 10.1016/j.critrevonc.2016.02.001
- Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013; 39(1): 1-10. doi: 10.1016/j.immuni.2013.07.012
- Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016; 16(5): 275-287. doi: 10.1038/nrc.2016.36
- Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012; 366(26): 2443-2454. doi: 10.1056/NEJMoa1200690
- Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014; 515(7528): 563-567. doi: 10.1038/nature14011
- Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014; 371(23): 2189-2199. doi: 10.1056/NEJMoa1406498
- Buchbinder EI, Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016; 39(1): 98-106. doi: 10.1097/COC.0000000000000239
- Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T-cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010; 107(9): 4275-4280. doi: 10.1073/pnas.0915174107
- Sliwkowski MX, Mellman I. Antibody therapeutics in cancer. Science. 2013; 341(6151): 1192-1198. doi: 10.1126/science.1241145
- Thompson RH, Kuntz SM, Leibovich BC. Tumor B7-H1 is associated with poor prognosis in renal cell carcinoma patients with long-term follow-up. Cancer Res. 2006; 66(7): 3381-3385. doi: 10.1158/0008-5472.CAN-05-4303
- Hamanishi J, Mandai M, Iwasaki M, et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci U S A. 2007; 104(9): 3360-3365. doi: 10.1073/pnas.0611533104
- Hino R, Kabashima K, Kato Y, et al. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer. 2010; 116(7): 1757-1766. doi: 10.1002/cncr.24899
- Hsu MC, Hsiao JR, Chang KC, et al. Increase of programmed death-1-expressing intratumoral CD8 T-cells predicts a poor prognosis for nasopharyngeal carcinoma. Mod Pathol. 2010; 23(10): 1393-1403. doi: 10.1038/modpathol.2010.130
- Mu CY, Huang JA, Chen Y, Chen C, Zhang XG. High expression of PD-L1 in lung cancer may contribute to poor prognosis and tumor cells immune escape through suppressing tumor infiltrating dendritic cells maturation. Med Oncol. 2011; 28(3): 682-688. doi: 10.1007/s12032-010-9515-2
- Sweis RF, Galsky MD. Emerging role of immunotherapy in urothelial carcinoma-Immunobiology/biomarkers. Urol Oncol. 2016; 34(12): 556-565. doi: 10.1016/j.urolonc.2016.10.006
- Fehrenbacher L, Spira A, Ballinger M, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016; 387(10030): 1837-1846. doi: 10.1016/S0140-6736(16)00587-0
- Chen J, Jiang CC, Jin L, Zhang XD. Regulation of PD-L1: a novel role of pro-survival signalling in cancer. Ann Oncol. 2016; 27(3): 409-416. doi: 10.1093/annonc/mdv615
- Liu L, Mayes PA, Eastman S, et al. The BRAF and MEK Inhibitors Dabrafenib and Trametinib: Effects on Immune Function and in Combination with Immunomodulatory Antibodies Targeting PD-1, PD-L1, and CTLA-4. Clin Cancer Res. 2015; 21(7): 1639-1651. doi: 10.1158/1078-0432.CCR-14-2339
- Toomer KH, Chen Z. Autoimmunity as a double agent in tumor killing and cancer promotion. Front Immunol. 2014; 5: 116. doi: 10.3389/fimmu.2014.00116
- Weber JS, OʼDay S, Urba W, et al. Phase I/II study of ipilimumab for patients with metastatic melanoma. J Clin Oncol. 2008; 26(36): 5950-5956. doi: 10.1200/JCO.2008.16.1927
- Weber J, Thompson JA, Hamid O, et al. A randomized, double-blind, placebo-controlled, phase II study comparing the tolerability and efficacy of ipilimumab administered with or without prophylactic budesonide in patients with unresectable stage III or IV melanoma. Clin Cancer Res. 2009; 15(17): 5591-5598. doi: 10.1158/1078-0432.CCR-09-1024
- Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: A randomized, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010; 11(2): 155-164. doi: 10.1016/S1470-2045(09)70334-1
- OʼDay SJ, Maio M, Chiarion-Sileni V, et al. Efficacy and safety of ipilimumab monotherapy in patients with pretreated advanced melanoma: A multicenter single-arm phase II study. Ann Oncol. 2010; 21(8): 1712-1717. doi: 10.1093/annonc/mdq013
- Hersh EM, OʼDay SJ, Powderly J, et al. A phase II multicenter study of ipilimumab with or without dacarbazine in chemotherapy-naïve patients with advanced melanoma. Invest New Drugs. 2011; 29(3): 489-498. doi: 10.1007/s10637-009-9376-8
- Boutros C, Tarhini A, Routier E, et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat Rev Clin Oncol. 2016; 13(8): 473-486. doi: 10.1038/nrclinonc.2016.58
- Kumar V, Chaudhary N, Garg M, Floudas CS, Soni P, Chandra AB. Current Diagnosis and Management of Immune Related Adverse Events (irAEs) Induced by Immune Checkpoint Inhibitor Therapy. Front Pharmacol. 2017; 8: 49. doi: 10.3389/fphar.2017.00049
- Lipson EJ, Drake CG. Ipilimumab: An anti-CTLA-4 antibody for metastatic melanoma. Clin Cancer Res. 2011; 17(22): 6958-6962. doi: 10.1158/1078-0432.CCR-11-1595
- Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015; 373: 23-34. doi: 10.1056/NEJMoa1504030
- Gao J, Ward JF, Pettaway CA, et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat Med. 2017; 23(5): 551-555. doi: 10.1038/nm.4308
- Koyama S, Akbay EA, Li YY, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016; 7: 10501. doi: 10.1038/ncomms10501
- Ludin A, Zon LI. Cancer immunotherapy: The dark side of PD-1 receptor inhibition. Nature. 2017; 552(7683): 41-42. doi: 10.1038/nature24759
- Wartewig T, Kurgyis Z, Keppler S, et al. PD-1 is a haploinsufficient suppressor of T-cell lymphomagenesis. Nature. 2017; 552(7683): 121-125. doi: 10.1038/nature24649
- Ribas A, Hamid O, Daud A, et al. Association of Pembrolizumab With Tumor Response and Survival among Patients with Advanced Melanoma. JAMA. 2016; 315(15): 1600-1609. doi: 10.1001/jama.2016.4059
- Diem S, Kasenda B, Spain L, et al. Serum lactate dehydrogenase as an early marker for outcome in patients treated with anti-PD-1 therapy in metastatic melanoma. Br J Cancer. 2016; 114(3): 256-261. doi: 10.1038/bjc.2015.467
- Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell. 2017; 168(4): 707-723. doi: 10.1016/j.cell.2017.01.017
- Calapre L, Warburton L, Millward M, Ziman M, Gray ES. Circulating tumour DNA (ctDNA) as a liquid biopsy for melanoma. Cancer Lett. 2017; 404: 62-69. doi: 10.1016/j.canlet.2017.06.030
- Lipson EJ, Forde PM, Hammers HJ, Emens LA, Taube JM, Topalian SL. Antagonists of PD-1 and PD-L1 in cancer treatment. Semin Oncol. 2015; 42(4): 587-600. doi: 10.1053/j.seminoncol.2015.05.013
- Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single-agent anti–programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010; 28(19): 3167-3175. doi: 10.1200/JCO.2009.26.7609
- Jamieson NB, Maker AV. Gene-expression profiling to predict responsiveness to immunotherapy. Cancer Gene Ther. 2017; 24(3): 134-140. doi: 10.1038/cgt.2016.63
- Le DT, Uram JN, Wang H, et al. Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015; 372(26): 2509-2520. doi: 10.1056/NEJMoa1500596
- Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015; 348(6230): 124-128. doi: 10.1126/science.aaa1348
- Hugo W, Zaretsky JM, Sun L, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016; 165(1): 35-44. doi: 10.1016/j.cell.2016.02.065
- Bronstein D. Who benefits the most from ipilimumab restricted distribution? Some question safety/REMS rationale. Pharm Pract News. 2011; 38(8).
- Sunshine J, Taube JM. PD-1/PD-L1 inhibitors. Curr Opin Pharmacol. 2015; 23: 32-38. doi: 10.1016/j.coph.2015.05.011
- Brahmer JR. Harnessing the immune system for the treatment of nonsmall-cell lung cancer. J Clin Oncol. 2013; 31(8): 1021-1028. doi: 10.1200/JCO.2012.45.8703
