

Research Article
NMR 1H Spectra of the [1,2]Diazepino[4,5-b]Indole Derivatives: Experimental versus GIAO calculated Data
1Department of the Biologically Active Compounds Chemistry, L.M. Litvinenko Institute of Physical Organic and Coal Chemistry R. Luxemburg str. 70, Donetsk, 283114, Ukraine
2Supramolecular Chemistry Depertment, L.M. Litvinenko Institute of Physical Organic and Coal Chemistry R. Luxemburg str. 70, Donetsk, 283114, Ukraine
*Corresponding author: Alexander Borisovich Eresko, Senior Research Fellow, L.M. Litvinenko Institute of Physical Organic Chemistry and Coal Chemistry, Donetsk, Ukraine, E-mail: a_eresko77@mail.ru
Elena Vladimirovna Raksha, Research Fellow, L.M. Litvinenko Institute of Physical Organic Chemistry and Coal Chemistry, Donetsk, Ukraine, E-mail: elenaraksha411@gmail.com
Received: December 25, 2018 Accepted: January 18, 2019 Published: January 28, 2019
Citation: Muratov AV, Berestneva YV, Zinchenko SY, et al. NMR 1H Spectra of the [1,2]Diazepino[4,5-b]Indole Derivatives: Experimental versus GIAO calculated Data. Int J Chem Res. 2018; 1(1): 9-12. doi: 10.18689/ijcr-1000102
Copyright: © 2019 The Author(s). This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
A comprehensive study of new [1,2]diazepino[4,5-b]indole derivatives molecular geometry as well as their NMR 1H spectra by DFT method was performed. GIAO-calculated NMR 1H chemical shifts as obtained at B3LYP/6-31G (d,p)/PCM computational level are reported for the 1,2-diazepine compounds.
Keywords: [1,2]Diazepino[4,5-b]indoles; B3LYP; GIAO; NMR 1H spectra; Chemical shift.
Introduction
The biological activity of compounds containing in their structure the 1-aryl-2,3-benzodiazepine skeleton is best demonstrated by the example of tofisopam, a wellknown anxiolytic drug, or talampanel [1,2]. Related condensed heterocyclic systems, such as indolo-1,2-diazepines, can be considered as potentially useful structures for drug design. It is known that indole-condensed azacycles are structural components of many natural and synthetic biologically active compounds [3,4]. Investigations on the synthesis of indolo[1,2]diazepines are few [5-8] and limited to derivatives containing alkyl substituent’s in the diazepine ring. Synthesis and structural modification of arylcontaining indolodiazepines have been demonstrated recently [9]. As a continuation of our research on the synthesis and reactivity of heterocyclic systems based on 1,2-diazepine [8,10-12], this paper presents the results of molecular modeling of the structure and NMR 1H spectra of some new indolo[1,2]diazepines.
Experimental Part
NMR 1H spectra were recorded in DMSO-d6 on 400/100 MHz NMR spectrometer (Bruker Avance II 400) and chemical shifts values (δ) are given in parts per million relative to tetramethylsilane (TMS). The melting points were determined on a Boetius hot stage. The CHN elemental analysis was performed using a Varian MICRO Cube analyzer. The mass spectra were recorded on an Agilent 1100 LC/MSD VL instrument (atmospheric pressure chemical ionization; Zorbax SBC18 column, 50 × 4.6 mm; eluent acetonitrile-water (95:5) containing 0.1% of trifluoroacetic acid, flow rate 3.0 mL/min, gradient elution).
For detailed information on the synthesis of [1,2]diazepino[4,5-b]indoles 1-3 as well as their precursors, see our recent article [9].
1-(4-methylphenyl)-5,10-dihydro[1,2]diazepino[4,5-b]indol-4(3H)-one (1).
Yield 45%, M.p. 281-282°С. NMR 1H (DMSO-d6), δ, ppm: 2.42 s (3Н, 4′-СН3), 3.59 s (2Н, СН2), 7.06 t (1Н, Н-7, J 8.0 Hz), 7.18 t (1Н, Н-8, J 8.0 Hz), 7.24 d (2Н, Н-3′,5′, J 8.0 Hz), 7.38 d (1Н, Н-9, J 8.0 Hz), 7.65 d (3Н, H-6,2′,6′, J 8.0 Hz), 10.64 s (1Н, CONH), 11.08 s (1Н, NH). Mass-spectrum: m/z 290.2 [M + 1]+. Found, %: С 74.70; Н 5.25; N 14.51. Anal. Calcd. for C18H15N3O, %: С 74.72; Н 5.23; N 14.52. M 289.33.
1-(4-methylphenyl)-5,10-dihydro[1,2]diazepino[4,5-b]indole-4(3H)-thione (2).
Yield 82%, M.p. 200-202°С. NMR 1H (DMSO-d6), δ, ppm: 2.44 s (3Н, 4′-СН3), 4.01 s (2Н, СН2), 7.13 t (1Н, H-7, J 8.0 Hz), 7.26 t (1Н, H-8, J 8.0 Hz), 7.32 d (2Н, H-3′,5′, J 8.0 Hz), 7.42 d (1Н, H-9, J 8.0 Hz), 7.66 d (2Н, H-2′,6′, J 8.0 Hz), 7.73 d (1Н, H-6, J 8.0 Hz), 11.41 s (1Н, NH), 12.51 s (1Н, CSNH). Mass-spectrum: m/z 306 [M + 1]+. Found, %: С 71.02; Н 4.57; N 14.13. Anal. Calcd. for C18H15N3S, %: С 70.79; H 4.95; N 13.76. M 305.40.
1-(4-methylphenyl)-4-(pyrrolidin-1-yl)-5,10-dihydro[1,2]diazepino[4,5-b]indole (3a)
Yield 72%, M.p. 273-275°С (with decomposition). NMR 1H (DMSO-d6), δ, ppm: 1.90 s (4Н, NСН2СН2 pyrrolidine), 2.42 s (3Н, 4′-СН3), 3.00 br.s (1Н, СН2), 3.51 br.s (5Н, NСН2СН2 pyrrolidine, СН2), 7.03 t (1Н, H-7, J 8.0 Hz), 7.14 t (1Н, H-8, J 8.0 Hz), 7.23 d (2Н, H-3′,5′, J 8.0 Hz), 7.38 d (1Н, H-9, J 8.0 Hz), 7.65 d (1Н, H-6, J 8.0 Hz), 7.68 d (2Н, H-2′,6′, J 8.0 Hz), 11.09 s (1Н, NH). Mass-spectrum: m/z 343 [M + 1]+. Found, %: С 77.28; Н 6.31; N 16.47. Anal. Calcd. for C22H22N4, %: C 77.16; H 6.48; N 16.36. M 342.44.
1-(4-methylphenyl)-4-(morpholin-1-yl)-5,10-dihydro[1,2]diazepino[4,5-b]indole (3b)
Yield 62%, M.p. 221-223°С. NMR 1H (DMSO-d6), δ, ppm: 2.43 s (3Н, 4′-СН3), 2.98 br. s (1Н, СН2), 3.38 br. s (4Н, NСН2 morpholine), 3.61 br. s (4Н, O-СН2 morpholine),4.30 s (1Н, СН2), 7.04 t (1Н, H-7, J 8.0 Hz), 7.17 t (1Н, H-8, J 8.0 Hz), 7.24 d (2Н, H-3′,5′, J 8.0 Hz), 7.39 d (1Н, H-9, J 8.0 Hz), 7.70 d (3Н, H-6, H-2′,6′, J 8.0 Hz), 11.16 s (1Н, NH). Mass-spectrum: m/z 359 [M+1]+. Found, %: С 73.89; Н 5.97; N 15.84. Anal. Calcd. for C22H22N4O., %: C 73.72; H 6.19; N 15.63. M 358.43.
Theoretical Methods and Computational Details
Initial molecular geometries of the [1,2]diazepino[4,5-b]indoles 1-3 for molecular modeling were generated using the algorithm of complete inclusion of possible geometric and steric factors implemented in the Conformer plug-in of the Marvin software package [13]. This algorithm enables generation of molecular structures with complete analysis of the carbon skeleton, functional groups and heteroatom’s, geometric isomers, and asymmetric centers.
Molecular geometry and electronic structure parameters, thermodynamic characteristics of the [1,2]diazepino[4,5-b]indoles 1-3 conformers were calculated using the Gaussian 09 [14] software package. Geometric parameters, harmonic vibrational frequencies, and the vibrational contribution to the zero-point vibrational energy were determined after full geometry optimization in the framework of B3LYP/6-31G (d,p) density functional calculations. The solvent (DMSO) effect was considered in the PCM approximation [15]. The optimized geometric parameters were used for total electronic energy calculations. The 6-31G (d,p) basis set was used in this work because it has a low computational cost. Only the lowest energy conformers of the [1,2]diazepino[4,5-b]indoles 1-3 were used for further consideration.
The magnetic shielding tensors (χ, ppm) for 1H nuclei of the [1,2]diazepino[4,5-b]indoles 1-3 were calculated with the B3LYP/6-31G(d,p)/PCM optimized geometries by standard GIAO (Gauge-Independent Atomic Orbital) approach [16]. The calculated magnetic isotropic shielding tensors, χi, were transformed to chemical shifts relative to TMS, δi, by δi=χref-χi, where both, χref and χi, were taken from calculations at the same computational level. χ Values for magnetically equivalent nuclei were averaged.
Results and Discussion
The main method of a 1,2-diazepine ring condensed with a heterocyclic fragment formation is the condensation of 1,5-dicarbonyl compounds with hydrazine [8,10-12]. Our approach to obtaining [1,2]diazepino[4,5-b]indoles 1-3 involves the synthesis of the starting 1,2-diazepine 1 and its subsequent structural modification to obtain compounds 2-3 (Figure 1).
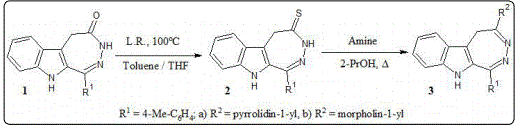
The starting [1,2]diazepino[4,5-b]indol-4-one 1 was obtained by cyclization of ethyl [2-(4-methylbenzoyl)-1H-indole-3-yl] acetic acid with hydrazine in the presence of a catalytic amount acetic acid. Then [1,2]diazepino[4,5-b]indol-4-one 1 was transformed to the corresponding thion 2 by reaction with Lawesson’s reagent. Diazepin-4-thion 2 interacts with corresponded amine when heated in propanol-2 with the formation of compounds 3a,b. The structure of the obtained new compounds 1-3 was confirmed by NMR spectroscopy.
The peculiarity of the NMR 1H spectrum of N-substituted [1,2]diazepino[4,5-b]indol-4-ones 3a,b compared with the corresponding 1,2-diazepin-4-one 1 and 1,2-diazepin-4-thion 2 is the appearing of the methylene group protons as two broadened singlets with chemical shifts of 3.00 and 3.51 ppm (compound 3a) as well as 2.98 and 4.30 ppm (compound 3b), which indicates a non-planar configuration of the diazepine ring. It is obvious that the presence of two large substituent’s in the diazepine ring reduces its conformational mobility, thus in the NMR 1H spectrum of compound 3, separate signals correspond to the equatorial and axial protons of the methylene group.
For the studied [1,2]diazepino[4,5-b]indoles 1-3, molecular geometry optimization was performed in the B3LYP/6-31G(d,p)/PCM approximation (solvent is dimethyl sulfoxide). The parameters of their molecular geometry and electronic structure were estimated. Some of the characteristics obtained are listed in table 1. For all the 1,2-diazepins mentioned, only conformers with the lowest total energy were considered. Structural models of 1,2-diazepines 1-3 with the numbering of atoms used in the discussion of the calculated chemical shifts, are shown on figure 2.
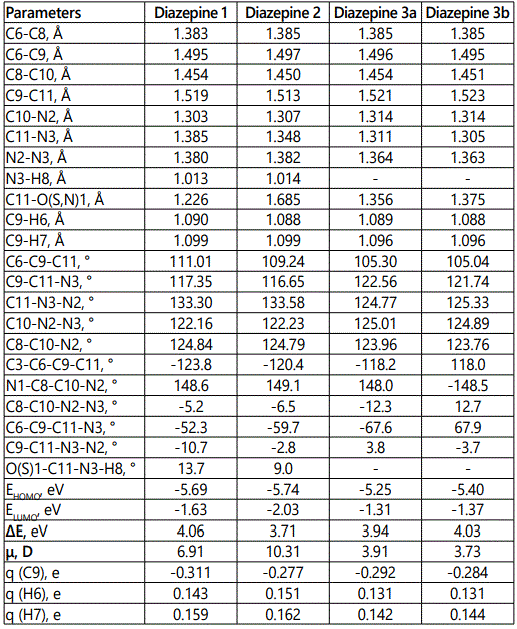
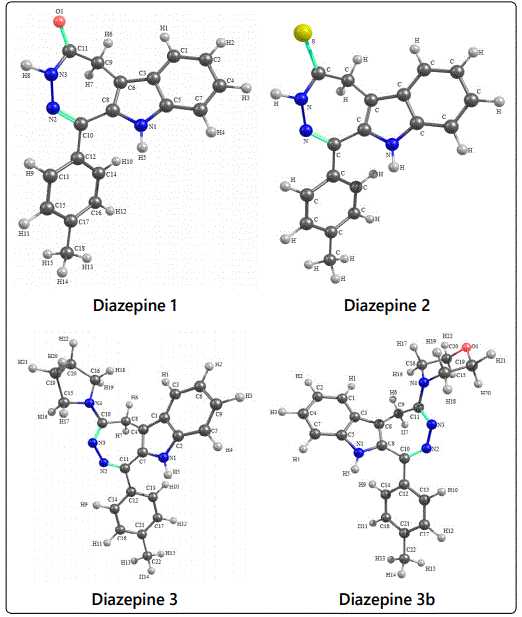
For the most stable conformers of the studied 1,2-diazepines molecules, the NMR 1H chemical shifts were estimated. To calculate the magnetic shielding constants using the standard GIAO method, the equilibrium configurations of compounds 1-3, obtained by B3LYP/6-31G(d,p) method with PCM approximation were used. The obtained chemical shifts for the compounds 1-3 are given in table 2. The NMR 1H parameters of the studied 1,2-diazepines are correctly reproduced at this theoretical level except for NH protons. It should be noted that taking into account non-specific solvation within PCM model is not sufficient for the correct reproduction of these protons, and the formation of hydrogen bonds with the solvent molecules should be considered. Thus chemical shifts of NH protons were not considered in further discussion. Linear relationships between the experimental chemical shifts and the calculated ones have been obtained for all studied diazepines molecules (Figure 3). The correlation coefficients (R) corresponding to obtained dependences are within 0.993–0.999. Equations, obtained for the individual compounds and the total one:
1. δexp=(0.952 ± 0.009)·δcalc+(0.14 ± 0.06), R=0,99946;
2. δexp=(0.953 ± 0.007)·δcalc+(0.15 ± 0.05), R=0,99966;
3a. δexp=(0.952 ± 0.037)·δcalc+(0.09 ± 0.22), R=0,99317;
3b. δexp=(0.945 ± 0.015)·δcalc+(0.18 ± 0.05), R=0,99735;
Total: δexp=(0.945 ± 0.013)·δcalc+(0.18 ± 0.08), R=0,99628.
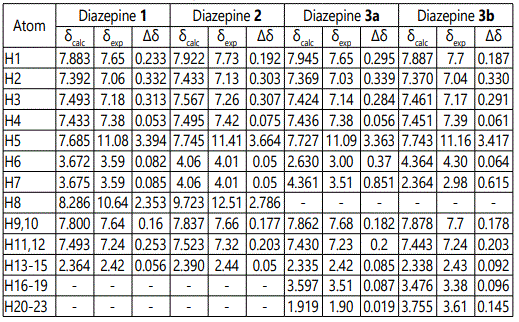

For N-substituted [1,2]diazepino[4,5-b]indoles 3a and 3b difference between calculated chemical shifts for non-equivalent methylene group protons exceeds the experimental value. This is due to the fast dynamics of the diazepine ring in the NMR time scale. With an increase in the temperature of the NMR experiment, one can expect higher conformation exchange rates, and, as a consequence, an even greater closing-in of these protons signals up to their coalescence. We observed such pattern for the 1,4-biaryl derivatives of benzofuro[2,3-d] [1,2]-diazepines experimental NMR 1H spectra [17]. Thus, a comprehensive study of the 1,2-diazepine core dynamics of condensed diazepines by dynamic NMR spectroscopy as well as DFT method will be the next stage of our work.
Conclusion
A comprehensive study of the [1,2]diazepino[4,5-b]indole derivatives by experimental NMR 1H spectroscopy and molecular modeling methods was performed. Structural parameters of the studied diazepines compounds were obtained by B3LYP method. GIAO-calculated NMR 1H chemical shifts as obtained at B3LYP/6-31G (d,p)/PCM computational level are reported for the [1,2]diazepino[4,5-b]indoles. For NMR 1H spectra of the diazepines in DMSO-d6 this method approximation allows to obtain the correct spectral pattern. Linear correlations between the calculated and experimental values of the 1H chemical shifts for the studied molecules were obtained.
References
- Luszczki JJ. Third-generation antiepileptic drugs: mechanisms of action, pharmacokinetics and interactions. Pharmacol Rep. 2009; 61(2): 197–216. doi: 10.1016/S1734-1140(09)70024-6.
- Iwamoto FM, Kreisl TN, Kim L, et al. Phase 2 trial of talampanel, a glutamate receptor inhibitor, for adults with recurrent malignant gliomas. Cancer. 2010; 116(7): 1776–1782. doi: 10.1002/cncr.24957.
- Sundberg RJ. Indoles. Academic: New York NY. 1996.
- Somei M, Yamada F. Simple indole alkaloids and those with a nonrearranged monoterpenoid unit. Nat Prod Rep. 2005; 22(1): 73-103. doi: 10.1039/b316241a
- Monge Vega A, Martinez MT, Palop JA, Mateo JM, Fernandez-Alvarez EJ. Synthesis of 1H-[1,2]diazepino[4,5-b]indole derivatives. Heterocyclic Chem. 1981; 18(5): 889-892. doi: 10.1002/jhet.5570180508
- Monge A, Palop JA, Goni T, Martinez AJ. Synthesis of 3H[1,2]diazepino[5,6-b]indole and 3H[1,2]diazepino[4,5-b]indole derivatives. Heterocyclic Chem. 1984; 21(2): 381-384. doi: 10.1002/jhet.5570210221
- Hatzimimikou D, Livadiotou D, Tsoleridis CA, Stephanidou-Stephanatou J. One-Step Synthesis of [1,2]Diazepino[4,5-b]indole Derivatives from the Reaction of Pyranoindolones with Methylhydrazine. Synlett. 2008; (12): 1773-1776. doi: 10.1055/s-2008-1078511
- Tolkunov VS, Eresko AB, Khizhan AI, Shishkin OV, Palamarchuk GV, Tolkunov SV. Heterocyclization of 2-acyl-3-indolylacetic acids using hydrazine. Synthesis of 2,3-dihydro-2-oxo-5-R1-1H-[1,2]diazepino[4,5-b]indoles. Chem Heterocycl Comp. 2009; 45(6): 726-734. doi:10.1007/s10593-009-0322-7
- Eresko AB, Tolkunov VS, Tolkunov SV. Condensed diazepines. Synthesis of 1-aryl-3,5-dihydro-4H-1-benzofuro-[2,3-d][1,2]diazepin-4-ones. Chem Heterocycl Comp. 2010; 46(9): 1127-1132. doi: 10.1007/s10593-010-0637-4
- Wang J, Wang L, Guo S, Zha S, Zhu J. Synthesis of 2,3-Benzodiazepines via Rh(III)-Catalyzed C-H Functionalization of N-Boc Hydrazones with Diazoketoesters. Org Lett. 2017; 19(13): 3640-3643. doi: 10.1021/acs.orglett.7b01642
- Espahbodinia M, Ettari R, Wen W, et al. Development of novel N-3-bromoisoxazolin-5-yl substituted 2,3-benzodiazepines as noncompetitive AMPAR antagonists. Bioorg Med Chem. 2017; 25(14): 3631-3637. doi: 10.1016/j.bmc.2017.05.036
- ChemAxon. Marvin 5.10.4. Calculator Plugins. 2014.
- Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 09, Revision B.01. Gaussian, Inc., Wallingford CT. 2010.
- Mennucci M, Tomasi J. Continuum solvation models: A new approach to the problem of solute’s charge distribution and cavity boundaries. J Chem Phys. 1997; 106(12): 5151-5158. doi: 10.1063/1.473558
- Wolinski K; Hilton JF; Pulay P. Efficient implementation of the gaugeindependent atomic orbital method for NMR chemical shift calculations. J Am Chem Soc. 1990; 112(23): 8251-8260. doi: 10.1021/ja00179a005
- Muratov AV, Grebenyuk SA, Eresko AB. Synthesis of 1,2-Diazepines by the Bischler–Napieralski Reaction. Russ J Org Chem. 2018; 54(6): 861-866. doi: https://doi.org/10.1134/S1070428018060064
