

Research Article
Pharmaceutical Bioinformatics: Its Relevance to Drug Metabolism
1 Faculty of Pharmaceutical Sciences, University of Nigeria, Nsukka, Nigeria
2 Faculty of Pharmaceutical Sciences, Enugu State University of Sciences and Technology, Nigeria
*Corresponding author: Chika John Mbah, Professor, Faculty of Pharmaceutical Sciences, University of Nigeria, Nsukka, Nigeria, E-mail: chika.mbah@unn.edu.ng
Received: December 19, 2018 Accepted: December 27, 2018 Published: January 7, 2019
Citation: Mbah CJ, Okorie NH. Pharmaceutical Bioinformatics: Its Relevance to Drug Metabolism. Madridge J Bioinform Syst Biol. 2018; 1(1): 19-26. doi: 10.18689/mjbsb-1000104
Copyright: © 2018 The Author(s). This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Bioinformatics as it relates to medicine involves the processing of the genetic information with the hope of generating the genetic basis of health and disease that could result in the efficient discovery of tailored and targeted drugs. Pharmaceutical bioinformatics therefore, deals with research problems requiring biological-sequence data, important sources of information, methods of access and the role of libraries and information centers as they relate to drug discovery, development and biotransformation. Drug biotransformation (metabolism) gives metabolites with physicochemical and pharmacological properties that differ significantly from those of the parent drug. It is usually investigated by experimental and computational approaches. Due to the importance of drug metabolism in terms of safety and efficacy, it becomes imperative to have efficient and reliable ways to predict drug metabolism in vitro, in silico, and in intact organisms. Molecular modeling and data modeling are in silico tools available for predicting drug metabolism. Prediction of drug metabolism has applications in drug design, medicinal chemistry, pharmacokinetics, toxicology and helps in the structural characterization of metabolites. The present study gives a comprehensive review of bioinformatics, biological processes (DNA and protein sequences), biological databases, search tools and similarity searching. The study also considered pharmaceutical bioinformatics and its application to drug metabolism.
Keywords: Bioinformatics; Pharmaceutical Bioinformatics; Prediction of Drug Metabolism.
Introduction
Bioinformatics is a branch of science that incorporates biology, computer science and information technology. It involves collection, organization, analysis, manipulation, presentation and distribution of biological data to help solve biological problems on the molecular level using computer technology. Its basic objectives involve data management and knowledge discovery through amalgamation of computers, statistics and molecular biology. As an interface between modern biology and informatics, it entails discovery, development and implementation of computational algorithms and software tools in an effort to facilitate an understanding of the biological processes [1,2]. Biological processes occur in cells. Cells possess a central core known as the nucleus that is the store house of a vital molecule called DNA. The DNA molecules are packaged in units called chromosomes. The chromosomes and DNA are together known as genome. The genomes have specific regions called genes that spread throughout the genomes. The RNA likewise contain information however, their major function is to copy information from DNA selectively and travels to protein production sites where the information is translated into proteins. Proteins are built out of functional units known as domains (or motifs) and the domains have conserved sequence [3]. The biological process is presented in figure 1.
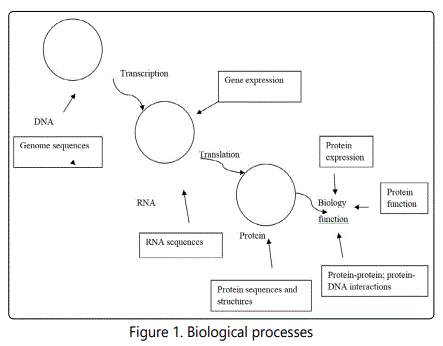
Classification of Bioinformatics
Bioinformatics classification could be based on (i) development and implantation of tools that will allow different types of information to be effectively and efficiently accessed and managed (databases). The development of bioinformatics tools is governed by the following biological processes: (a) DNA sequence- determines protein sequence (b) protein sequence- determines structure (c) protein structuredetermines protein function [4], (ii) analysis and interpretation data from various sources such as nucleotide and amino acids sequences, protein domains and protein structure (search tools). (iii) development of new algorithm and statistics in order to assess relationships among numbers of large data sets (similarity searching) [5].
Premier databases
As a result of the large volume of data that has been
produced, its organization and storage becomes necessary.
Thus, databases that constitute a large number of biological
information were created, stored, processed and provide
access to scientists [6]. National Center for Biotechnology
Information (NCBI) is the world's premier website for
biomedical and bioinformatics research. It was established in
1998 as national (USA) resource for molecular biology
information. NCBI creates public databases, carries out
research in computational biology, analyzes genome data
using in-house developed software tools and provides
understanding of molecular processes affecting human health
and diseases through dissemination of biomedical
information. Its service units include PubMed (the
bibliographic database), Gen Bank [nucleotide sequence
database, protein sequences, short RNA fragments (ESTs),
cancer genome anatomy project (CGAP) - gene expression
profiles of normal, pre-cancer, and cancer cells from a wide
variety of tissue types, single nucleotide polymorphisms
(SNPs) - which represent genetic variations in the human
population and online mendelian inheritance in man (OMIM)
- a database of human genetic disorders]. Each sequence in GenBank has a unique "accession number". The other world
premier databases are DNA Data Bank of Japan (DDBJ),
European Bioinformatics Institute (EBI) and European
Molecular Biology Laboratory (EMBL).
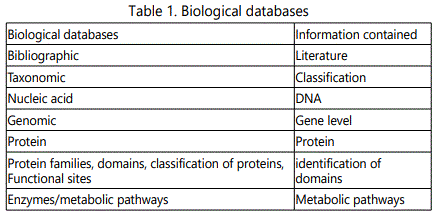
Biological databases
Biological databases (Table 1) are huge databases that
assist scientists to understand and explain biological
phenomena such as structures of biomolecules and their
interactions; metabolism of organisms and evolution of
species [7].
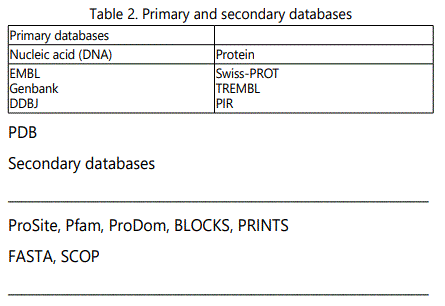
The biological databases (Table 2) are categorized into primary databases (contain sequence data for DNA, protein) and secondary databases (contain results from the analysis of the sequences in the primary databases). The primary databases are members of the International Nucleotide Sequence Database Collaboration (INSDC) and transfer the deposited information daily among each other. The secondary databases are curated and present only information related to proteins, describing aspects of its structure, domains, function, and classification. Information about DNA, proteins, protein functions normally stored in an intelligent fashion (databanks) enable scientists solve problems easily and quickly.
Such databanks include:
(a) Protein structure: Protein Databank (PDB)
(b) Protein sequence and their functions: Swiss-Port
(c) Interaction about enzymes and their functions: ENZYME
(d) Nucleotide sequences of all genes: EMBL
Employing databanks, all kinds of comparisons and search queries can be carried out [8,9].
Search tools
Entrez is the text-based search and retrieval system used
by NCBI for all the major databases such as PubMed (provides
access to citations including abstracts, full-text journal
articles), nucleotide and protein sequences, protein structures,
taxonomy etc. Entrez is much more than a tool for finding
sequences by keywords, it can also search for keywords such
as gene names, protein names, and the names of organisms
or biological functions. Entrez is internally cross-linked. For
instance, (i) DNA and protein sequences are linked to other
similar sequences
(ii) 3-D structures are linked to similar structures (iii) Medline (bibliographic database coverings fields of medicine, dentistry, nursing veterinary medicine etc.) citations are linked to other citations (PubMed) that contain similar keywords. This potential for horizontal movement through the linked databases makes Entrez a dynamic search and retrieval tool. Other search tools are PAM matrix (proteins), RasMol (simplest PDB viewer) etc.
Similarity searching
Consists of, a variety of computer programs used to make
comparisons between DNA sequences. BLAST (Basic Local
Alignment Search Tool) is complex and the most popular. It
generates an E-value for every match - (the same as the P
value in a statistical test). A match is generally considered
significant if the E-value<0.05 that is smaller numbers are
considered to be more significant. Similarity searching relies
on the concepts of alignment and distance between pairs of
sequences. Distances can only be measured between aligned
sequences for example match versus. Mismatch at each
position.
BLASTX makes automatic translation and allows DNA query sequence to compare with protein databanks, while TBLASTN makes automatic translation of an entire DNA database and allows it to be compared with protein query sequence [10].
Pharmaceutical Bioinformatics
Bioinformatics is of importance to Pharmacy (Pharmaceutical bioinformatics) in the areas of (i) drug discovery, designing and development, (ii) product/formulation designing, (iii) Pharmacokinetics and pharmacology. Pharmaceutical bioinformatics deals with scientific area of computer based technologies and informatics, computational methods for mapping processes of the cells (genetic information) and understanding how to use these properties to effectively discover and develop novel drugs. The novel drugs could be tailored or targeted drugs. Target drugs are drugs designed specifically to act on particular genes and their corresponding protein identified to be responsible for certain disease conditions. While tailored drugs refer to drugs designed to handle the needs of a specified genetic sub-group of the entire population [11,12]. The discovery and development process involve the employment of computer-aided drug design (CADD) methods. CADD methods are dependent on bioinformatics tools, applications and databases. The methods entail building three dimensional (3-D) virtual compound libraries (databases) for in silico screening (virtual screening) by docking the compounds against validated drug targets, followed by judicious selection of virtual hits possessing appropriate physicochemical properties to be screened for biological activity [13-15]. Some libraries consist of compounds with activities against several diseases, e.g. the ZINC database [16] while others are activity focused libraries [17]. The library is usually filtered to eliminate irrelevant molecules through a concept referred to as 'rapid elimination of swill' (REOS) [18]. REOS aids to identify molecules with poor absorption, distribution, metabolism, elimination and toxicology (ADME/T) properties. Thereafter, virtual screening is carried out by docking the "filtered out" library (or dataset) against validated drug targets in order to identify promising hit compounds, which are then subjected to biological activity assays.
Drug Metabolism and Enzymes
Elimination of drugs from the body occurs either by the process of excretion (unchanged), or conversion to metabolites with lower affinity characteristics (biotransformation). The biotransformation (metabolism) of a drug substance is the process whereby human beings effect chemical changes to a drug molecule and the product of such a chemical change is termed a drug metabolite [19-22].
Biotransformation is very significant in drug discovery and development due to the formation of active metabolites from active drugs; active metabolites from prodrugs (activation) and inactive metabolites (inactivation); toxic metabolites (toxification), metabolites that can inhibit metabolic pathway(s), metabolites that have physicochemical properties quite different from the parent compound (s) and producing complex kinetics.
Drug metabolism is one of the four discrete processes in the pharmacokinetic phase during the biological disposition of a drug. Drug metabolism reactions are classified as either phase I (functionalization reactions), or phase II, (biosynthetic (conjugation) reactions [23,24].
In Phase I reactions (oxidation, reduction, hydrolysis) functional group (s) is introduced on the parent compound, generally resulting in loss of pharmacological activity; but, active and chemically reactive intermediates could also be generated. Oxidation (most common) includes aromatic hydroxylation, deamination of mono- and diamines, dehydrogenations, N-, O-, and S-dealkylation, side-chain hydroxylation and sulphoxide formation. Reduction includes reduction of nitro, nitroso and azo groups while hydrolysis is the biotransformation route for esters and amides. In Phase II conjugation reactions (biosynthetic process), a covalent bond is formed between a functional group on the parent compound (or on a phase I metabolite) with endogenously derived glucuronic acid, sulphate, glutathione, amino acids or acetate. These conjugates are highly polar (generally inactive) and are rapidly excreted in the urine and faces. Drug metabolism takes place principally in the liver, however, other organs or tissues like the kidney, intestine, skeletal muscle, or even plasma could be important sites of metabolism. Most drug metabolism in a given cell occurs in the endoplasmic reticulum or cytosol, mitochondria, nuclear envelope and plasma membrane. Drug metabolisms are catalyzed by enzymes. The most important group of drug metabolizing enzymes is the Cytochrome P450 (monooxygenase system). Hydrolytic enzymes include a number of non-specific esterases and amidases (located in the endoplasmic reticulum of human liver, intestine and other tissues). The microsomal epoxide hydrolase considered a detoxification enzyme is present in the endoplasmic reticulum of essentially all tissues. It hydrolyzes highly reactive arene oxides (generated from CYP450 oxidation reactions) to inactive, water-soluble transdihydrodiol metabolites. The most important of conjugation enzymes are uridine diphosphate glucuronosyltransferases ('UGTs', microsomal enzymes), catalyzing the transfer of glucuronic acid to aromatic and aliphatic compounds. Other important enzymes involved conjugation reactions are sulphotransferases and N-acetyltransferases. Drug metabolism is currently being integrated into drug design and lead optimization strategies in order to reduce the cost and time taken to develop active compounds that might ultimately not be clinically successful due to hidden pharmacokinetic or toxicological defects [25].
In silico metabolism screening
One of the major fields within pharmaceutical
bioinformatics is the in silico metabolism prediction of drug
candidates [26]. It involves (i) predicting the occurrence of an
interaction between a compound and an enzyme, (ii)
predicting the location in the compound that takes part in the
interaction (the site of metabolism, SOM), (iii) predicting the
outcome from the interaction (the resulting metabolite
product). In metabolic prediction, scientists would like to
know (a) all reasonable phase I and phase II metabolites (b)
probability of formation under different biological conditions
(c) probability of formation based on molecular factors and a
filter against improbable metabolites (d) reactive/adductforming metabolites and itemize the metabolites. The
challenges facing reliable drug metabolism prediction include
(i) inter-individual factors (remain invariable for a given
organism) namely animal species, genetic factors, gender, (ii)
intra-individual factors (vary for a given organism) namely
age, biological rhythms, disease, stress, pregnancy, nutrition,
influence of inducers and inhibitors, (iii) selectivity
characteristics of metabolic processes for example one type
of selectivity at the receptor level (quantitatively or qualitatively
different responses elicited by various drug substances while
two different types of selectivity in drug metabolism (substrate
selectivity and product selectivity). Substrate selectivity is the
differential metabolism of distinct substrates under identical
conditions whereas product selectivity is the differential
formation of distinct metabolites from a single substrate
under identical conditions. Both types of selectivity can be
grouped into subtypes depending whether substrates (or
products) are non-isomeric (analogs, homologs or congeners),
regioisomeric (positional isomers), or stereo isomeric
(diastereomers or enantiomers). Both substrate and product selectivity are very vital in order to predict biotransformation.
In Silico systems to predict metabolism
A wide range of computational methods and integrated
approaches are used for the prediction of drug metabolism.
Molecular modeling and data modeling are in silico tools
available for predicting drug metabolism. Molecular modeling
[27] requires having knowledge about the three-dimensional
(3D) structure of the protein. Data modeling is useful for
information built from only known substrates or inhibitors
when information on the three-dimensional (3D) structure of
the protein is not available. Based on this, computational
methods are generally classified in two categories: ligandbased approaches [28], which use the information of the
substrate (ligand); and (ii) structure-based approaches [29,30],
which use the information of the enzyme-substrate complex.
Furthermore, two types of algorithms namely specific (local
systems) and comprehensive (global systems) can be used to
predict drug metabolism.
(A) Specific (local) systems: apply to simple biological systems (single metabolic enzymes or single metabolic reactions) and are usually restricted to rather narrow chemical series. Such systems include (i) quantitative structure- metabolism relationships (QSMRs) based on structural and physicochemical properties. It deals with affinities, relative rates etc. The relationships could be linear, multilinear, multivariate etc [31], (ii) quantum mechanical calculations revealing correlations between rates of metabolic oxidation and energy barrier in cleavage of the target C-H bond. It deals with regioselectivity, mechanisms, relative rates etc [32], (iii) three-dimensional QSMRs (3D-QSMRs) methods yielding a partial view of the binding/catalytic site of a given enzyme as derived from the 3D molecular fields of a series of substrates or inhibitors. It deals with substrate behavior, relative rates, inhibitor behavior etc. The 3D-QSARs has amongst other methods, two important ones such as CoMFA (comparative molecular field analysis) and GRID/GOLPE etc. (iv) molecular modeling and docking
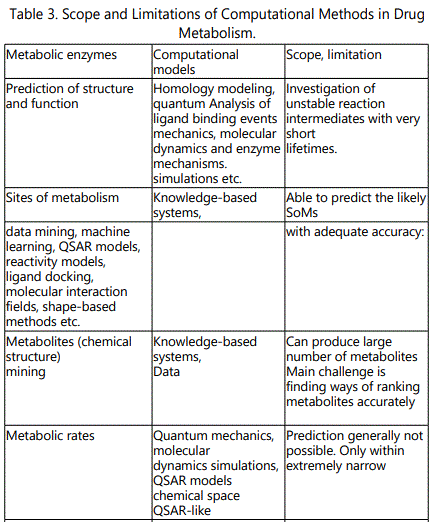
(B) Comprehensive (global) methods: apply to versatile biological systems (enzymes, reactions and/or series of compounds with broad chemical diversities. Such systems include: (i) Databases (MDL metabolite database, biotransformations etc). The databases deal with the nature of metabolites, reactive/adduct-forming metabolites etc. (ii) Expert systems and their databases (META, MetabolExpert, METEOR). They deal with the nature of major and minor metabolites, metabolic lists, reactive/adduct-forming metabolites, relative importance of these metabolites depending on biological factors etc. METEOR is a computer system which uses a knowledge base of structure-metabolism rules (biotransformations) to predict the metabolic fate of a query chemical structure. The reasoning model built into METEOR allows the system to evaluate the likelihood of a biotransformation taking place. The scope and limitation of computational methods in predicting drug metabolism is presented in table 3.
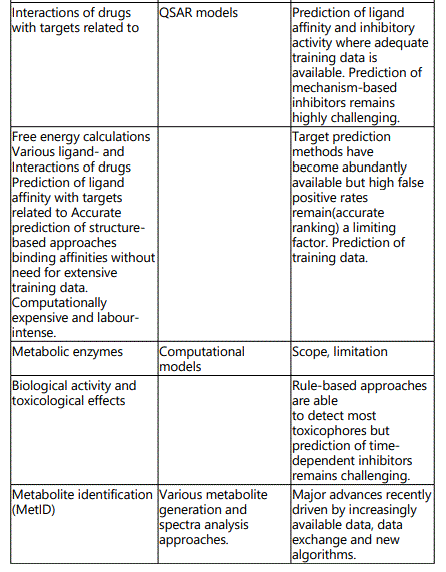
Outcome of prediction of drug metabolism
The successful prediction of drug metabolism depends
on data and information gathered from various methods and
resources. Such methods (models) and resources include:
Experimental data: Computational models are often (but
not exclusively) based on experimental data, and the amount and quality of the available data will determine their coverage
and performance. Experimental data such as bioactivities can
be modeled using QSAR techniques by applying linear regression
techniques to fit experimental data. Biotransformation data
can be used to derive models for predicting both the sites
and products of metabolism in an automated fashion. For
instance, MetaPrint2D [33] generates simple statistical models
for site of metabolism (SoM) prediction from biotransformation
databases. A modified form (MetaPrint2D-React of the
software, identifies and encodes the type of metabolic
reaction observed for specific atom environments and
generates the chemical structures of likely metabolites by
applying reaction rules to predicted site of metabolisms
(SoMs).
Expert knowledge: Scientists using empirical knowledge accumulated from drug metabolism research data developed reasoning models and have applied them to metabolite structure prediction [34]. Knowledge-based approaches such as Meteor [35] predict the sites and products of metabolism by scrutinizing a molecule of interest for the presence of target fragments. Their key advantage is the provision of the rational basis underlying a prediction (for example literature references and brief descriptions).
Physicochemical properties: Expert systems and many other predictors make extensive use of computed physicochemical properties such as logarithm partition coefficient (octanol/ water) or logarithm distribution coefficient (log D) and the knowledge that highly water-soluble compounds are likely to be excreted without undergoing metabolism as a means of metabolite ranking and filtering.
Target Structure: Consist of ligand-based and structurebased methods. Ligand-based has significant uncertainty about the target structure, specifically the ligand-receptor interaction site. Automated ligand docking can be utilized to examine if a specific site on a molecule has the potential to bind to a specific site in a target protein. It is possible to predict SoMs by relating the proximity of ligand atoms in a computed docking pose to the catalytic center of the target enzyme. This approach provides a structural hypothesis for the observed biological response and can correctly predict the approximate ligand orientation within the binding pocket [36,37]. To identify the SoM, a variety of ligand-based tools are used, such as expert systems, data mining approaches, quantitative structure activity relationships (QSAR), machinelearning methods, pharmacophore-based algorithms, shapefocused techniques, molecular interaction fields (MIFs), and reactivity-focused techniques. Structure-based methods consider structural properties of the target; these structural models cover only a fraction of the enzymes' conformational space relevant to the binding of small molecules [38].
Target Flexibility: The plasticity and size of drug-metabolizing enzymes binding sites depend on their functions and provide a flexible and adaptable system for processing a wide range of substrates. Molecular dynamics (MD) simulations/quantum chemical methods are the most powerful theoretical approaches for analyzing and predicting the interactions of protein-ligand pairs. Such simulation methods also provide knowledge about the structure, function, specificity and mechanisms of metabolic enzymes [39,40].
Reactivity: Quantum mechanical (QM) methods allow reactivity study. Reactivity is the major determinant of drug metabolism [41], QM systems generally consider only the most proximate protein environment (directly involved in a chemical reaction) but ignore effects originating from the more distant protein environment [42]. Molecular dynamics simulations and quantum mechanical methods have complementary properties and the combination has become a key technology for investigating enzyme reactions [43,44]. The calculation of molecular flexibility and/or reactivity, depict one specific protein-ligand interactions or enzyme mechanism only.
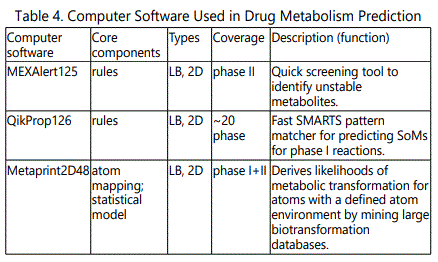
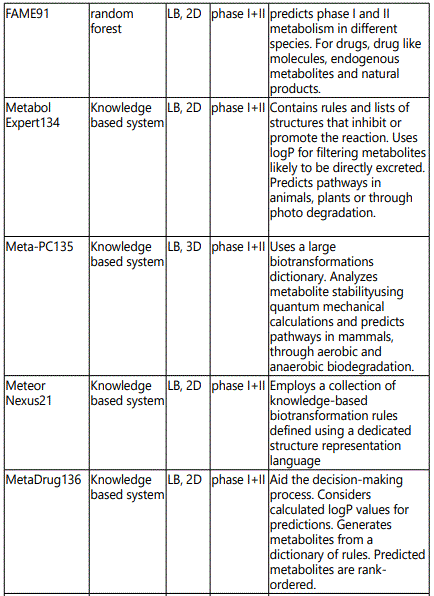
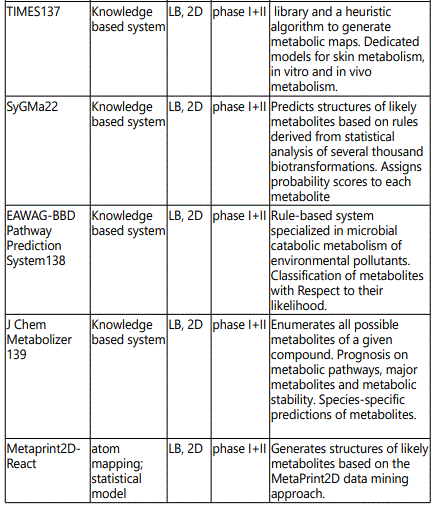
Metabolic networks - Systems biology: Comprehensive models (simulators) of drug metabolism require the ability to correctly predict various events and properties of the system to allow the estimation of biological effects. It would be accomplished by accurate knowledge and prediction of (a) concentrations and distribution of the drug, (b) metabolic liabilities (SoMs), (c) chemical structure of metabolites, (d) interactions with pharmacologically and toxicologically relevant biomolecules, (e) reaction rates and (f) tissue concentration and localization of enzymes and cofactors. Target prediction tools allow the identification of likely ligandprotein interactions and possibly extrapolation to the contribution of these interactions to prediction of phenotypic effects using QSAR techniques. QSAR models for predicting drug metabolism have undergone significant advances. The QSAR models can be divided into four main steps: (i) determination or collection of the biological property of interest (metabolism parameters), (ii) molecular descriptor generation and variable selection to extract desirable independent variables, (iii) model generation and validation with training and test sets using linear or nonlinear statistical methods, and (iv) prediction of the metabolism of new compounds using an external validation set. Several types of QSAR approaches have been developed with a wide variety of descriptors, such as: physicochemical (1D), topological (2D), and the 3D structure (3D) [45,46]. Table 4 shows the computer software utilized in predicting drug metabolism.
Conclusion
Experimental and integrated computational approaches have been used to investigate drug metabolism. Experimental approaches used to investigate drug metabolism come with substantial demands in technical resources and human expertise. Integrated computational approaches combine a variety of data sources, models, and algorithms in order to highlight applicability, information content and significance and prediction success rates with the major objective of rendering a complete picture of physiological processes.
Currently, research predicting drug metabolism has been limited to a number of technologies, namely rule-based tools and algorithms for sites of metabolism, electronic models, homology models as well as pharmacophores and QSARs models.
Due to the importance of human expertise, various disciplines such as chemistry (analytical, medicinal, physical, organic synthetic,), biology (biochemistry, enzymology, epigenetics, genetic etc.), pharmacology (clinical, molecular, pharmacokinetic, toxicology, therapeutics etc.), and computational components (software development, quantum chemistry, simulations, statistics, machine learning etc) are involved in drug metabolism prediction. Finally, the study has revealed the relevance of pharmaceutical bioinformatics in predicting and understanding drug metabolism (biotransformation) including information regarding the structure-metabolism relationships.
References
Contact us for any additional information - contact@madridge.org
 Madridge Publishers is licensed under a Creative Commons Attribution 4.0 International License.
Madridge Publishers is licensed under a Creative Commons Attribution 4.0 International License.