

Case Report
Vulvar Plexiform Neurofibromatosis, Case Report and Review of Literatures
Ain Shams University, Cairo, Egypt
*Corresponding author: Mohamed Ibrahim Mohamed Amer, Ain Shams University, Egypt, E-mail: mohamed_amer810@yahoo.com
Received: August 01, 2017 Accepted: August 10, 2017 Published: August 16, 2017
Citation: Amer MI, Alloob A. Vulvar Plexiform Neurofibromatosis, Case Report and Review of Literatures. Madridge J Womens Health Emancipation. 2017; 1(1): 7-10. doi: 10.18689/mjwh-1000103
Copyright: © 2017 The Author(s). This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Neurofibromatosis is a rare disease that can affects the female urogenital system
with tumor formation. Clitoral involvement is rare and selective labium majus
neurofibroma without clitoral involvement is extremely rare. Only 31 cases of
genitourinary neurofibromas have been described in the literature. We report on a
vulval plexiform neurofibroma of diffuse type in a child as a primary presentation of
neurofibromatosis without systemic manifestations of the disease nor clitoral involvement
which is extremely rare. Immunohistochemical study of the excised lesion is important
to confirm the diagnosis. Neurofibroma should be considered as one of the differential
diagnosis of the vulval swelling which would help in the primary surgery where extended
excision of the tumor is essential. Long-term follow-up is needed where there is increased
risk of recurrence and malignant transformation.
Keywords: Neurofibromatosis; Vulva; Tumor recurrence
Introduction
Plexiform neurofibroma is one type of the generalized neurofibromatosis. It is
considered to be a hamartoma rather than a typical tumor where it occurs due to
overgrowth of neural tissue in the subcutaneous fat [1]. The origin of this disease was
traced to a specific gene region on chromosome 17 which is autosomally dominant with
variable penetration [2]. Approximately 50% of patients have a family history of the
disorder; the lesions in the remainder are believed to arise from new mutations. The
frequency of malignant neurogenic tumors in patients with neurofibromatosis is higher
than in the general population, with rates varying from 5% to 30%, however, is rare in
children [3, 4]. The disorder usually is manifested in young children by cafe-au-lait spots,
where more than six spots are considered to be pathognomonic of the disease. Other
manifestations include bone overgrowth, congenital bowing and pseudarthrosis of
bone and soft tissue, kyphoscoliosis, focal gigantism, and megalencephaly [5, 6].
Clitoral neurofibromas in neurofibromatosis type 1 are rare. Clitoromegaly with
pseudopenis is the most frequent presentation of a neurofibroma in a female, while
labia majora neurofibroma without clitoral involvement is rare [7]. Less than 31 females
with NF1 with external genital involvement were recorded in the literatures [7, 8]. The
treatment of plexiform neurofibromas is by surgical excision, but complete removal is
difficult, being an infiltrating tumor, with an increased rate of recurrence [9]. The clinical
suspicion of NF might help in the management by preventing undue procedures [10].
Case
Premenarchal 11 years old girl presented by her mother
with a slowly growing, painless, right sided Vulval swelling of
few months duration. General examination revealed complete
female secondary sexual characters without any clinical
evidence of neurofibromatosis and there was no enlargement
of neither inguinal lymph nodes nor Liver and spleen.Local
examination revealed soft subcutaneous swelling with ill-
defined margins of 6 × 8 cm in diameters, located at right
labium majus, and attached to the overlying skin without
involvement of labia minora or clitoris.
ltrasound Duplex of the mass revealed an ill-defined
right vulval swelling measuring 49 × 71 × 30 mm presenting
hypoechoic center with echogenic peripheral portion. The
swelling is highly vascular presenting arterial channels showing
low resistance flow with a peak velocity of 31 cm/sec and
multiple venous channels inside. The swelling was at first
diagnosed as a case of haemangioma, lymphangioma or
lipoma, and excision biopsy was decided for final diagnosis.
At the operation, the mass was found very vascular
involving the epidermal and subcutaneous tissues with no line
of demarcation between the mass and the surrounding
apparently normal vulval tissues. Excision of the mass was
done with the overlying skin to the extent that both labia
majora became cosmetically of the same size. The excised
mass later proved to be plexiform neurofibromatosis at
histopathologic examination and confirmed by
immunohistochemical study. The patient warned of the
possibility of recurrence since the swelling was not completely
excised with a safety margin as proved at histopathologic
examination, and advised for follow up.
Four years later the patient presented with recurrence of
the Vulval swelling at the same site with the same criteria of
soft subcutaneous swelling with ill-defined edges and
attached to the overlying skin. Skin manifestations of
neurofibromatosis in the form of multiple cafe au lait spots
started to be manifested on the neck, chest, abdomen and
buttocks (Figure 1). Meticulous retaking of the family history
proved that her father having neurofibromatosis (Von
Recklinghausen's disease).
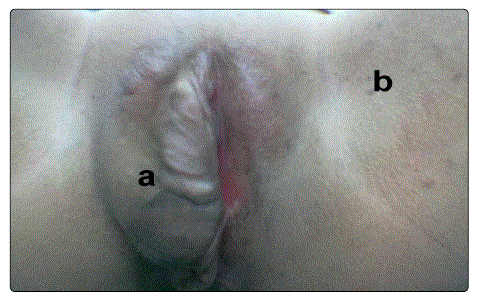
Figure 1: Right Vulval swelling due to plexiform neurofibroma of the diffuse type, b. inguinal freckling
Based on the previous diagnosis of plexiform
neurofibromatosis, decision for an extended excision of the
vulval swelling with a safety margin was taken. At the operation
the tissues were found very vascular and the swelling was
excised with a safety margin with extension to the labial fat
and perianal area.
Pathologic Findings
The specimen consisted of a grayish white mass of 8 x 4
cm size, firm in consistency with a covering ellipse of
corrugated skin, with a whitish whorly cut section.
Microscopically, within the dermis, there was a poorly
demarcated nodule composed of numerous anatomizing
fascicles of Schwann cells, that was separated by fibrous
bundles and extended among and replacing skin appendages.
Structures resembling pacinian corpuscles were also present.
Some of the anatomizing fascicles of Schwann cells showed
myxoid areas, whereas others exhibited fibrosis. Areas with a
diffuse pattern showing elongated Schwann cells with
serpentine, wavy nuclei were seen as well in Figures 2a & 2b.
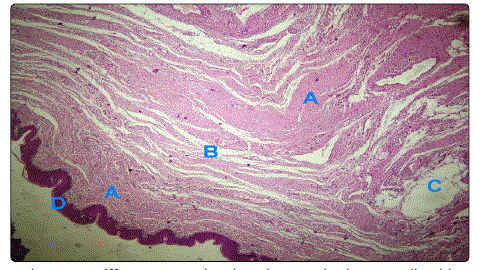
Figure 2a: Diffuse pattern showing elongated Schwann cells with serpentine, wavy nuclei

Figure 2b: Diffuse pattern showing elongated Schwann cells with serpentine, wavy nuclei
Immunohistochemistry with streptavidin-biotinperoxidase is positive for protein S-100 antibodies, where S-100 protein immunostain highlighted the fascicles of Schwann cells with areas exhibiting a diffuse pattern, confirming the diagnosis of plexiform neurofibroma of the vulva (Figure 3).
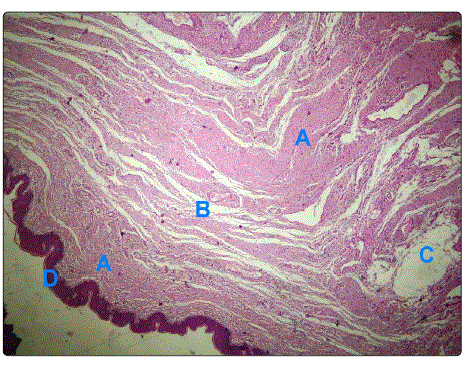
Figure 3.A. Anastomosing fascicles of Schwann cells, B. Fibrous
Bundles, C. Myxoid areas.
D. Vulval skin H&E, x100
Discussion
Genitourinary tract affection with NF1 in girls is rare where
most of it is presenting as clitoral hypertrophy. Of the 31 cases
recorded with Genitourinary neurofibroma in NF1, in 17
(57%), the neurofibroma only involved the clitoris; in 6 (20%),
the tumor also involved the bladder (in 2, the ureters also
were involved); and in 8 (27%), the neurofibroma also involved
the labia majora [7, 11-13].
In the present case, we describe a labium majus
involvement with plexiform neurofibroma of diffuse type in a
child as a primary presentation of NF1 without any other
clinical manifestations of the disease nor clitoral involvement
which is extremely rare [7].
Neurofibromatosis is a neuroectodermal heterogeneous
disease originating from the neural crest cells and is of two
types: NF1 (von Recklinghausen) and NF2 (acoustic neuroma).
The neurofibromatosis type 1 is an autosomal dominant
disease of incidence of 1 in 3,500 live births with spontaneous
mutations in half of all cases [14, 15]. It is caused by a gene on
chromosome 17, where its protein product (neurofibromin),
control cell growth and differentiation by tumor suppressor
action.
Mutation in the neurofibromatosis type 1 gene of neural
crest- derived cells is the molecular basis of genetic mosaicism,
and functional loss of neurofibromin in Schwann cells or
fibroblasts, determine the occurrence of neurofibromas or
pigmentary changes respectively [16]. Early mutations cause a
generalized disease whereas mutations occurring late in
embryogenesis give rise to an affected single region or organ
(localized disease) [16].
Genetic counseling is the first step in the management of
NF1, where if a parent is affected, the risk of NF for each
pregnancy is 50% and annual examinations in a
multidisciplinary clinic in the first 10 years of life are
recommended [17]. The diagnosis of most of cases of NF1 is
based on clinical manifestations in spite of great advances in genetic testing that diagnose as many as 95% of cases. The
prenatal diagnosis and diagnosis in patients with equivocal
physical signs as in a child with cafe au lait patches only could
be done with DNA analysis [17, 18].
To diagnose NF1, presence of 2 or more of the following
criteria is needed; 6 or more cafe au lait patches, axillary or
inguinal freckling, 2 or more cutaneous neurofibromas, 1
plexiform neurofibroma, characteristic bony lesions
(pseudarthrosis, sphenoid wing hypoplasia), an optic glioma,
2 or more iris Lisch nodules, or a first-degree relative with NF1
[19]. The diagnosis could be made at birth, while in others
observation for a few years is needed for the appearance of
additional criteria. Some patients have very mild manifestations,
whereas others are severely affected. Caféau-lait spots and
skin neurofibromas occur in 95% of patients or more, whereas
other criteria occur in less than 1% of cases, and consistency
in manifestations within a pedigree is not a rule [20].
Morbidity of NF1 could be in the form of disfiguring
cutaneous lesions, visual or hearing affection, pain, seizures,
spinal cord compression, bowel or urinary bladder
complications, Psychological stress. However, some of these
complications are infrequent, but psychosocial problems
remain a significant problem. Mental retardation, dysmorphic
facial features, and to a lesser extent early tumor burden is
more common in patients with large gene deletions, but, as
yet, there is no clear genotype/phenotype correlation [21].
The increased mortality in young adults could be attributed to
the increased incidence of cardiac diseases and malignant
tumors [22].
Cancer is 3% more in patients with neurofibromatosis
type 1 (NF1) than in general population with increased
frequency of unusual tumors as Carcinoid, pheochromocytoma,
brain tumors, chronic myeloid leukemia, and malignant
peripheral nerve sheath tumors [23]. Other tumors such as
breast, lung, kidney and colon, occurs less frequently than in
the normal population [24].
Neurofibromas, the hallmark of NF1, are tumors of the
autonomic peripheral nerves (Schwann cells) and rarely
involve organs in the urogenital system and may be found in
essentially any site, including the gastrointestinal tract, blood
vessels, heart, and larynx [15]. Plexiform neurofibroma (both
nodular and diffuse type) occurs in only 5 percent of patients
with NF1 [25]. Diffuse plexiform neurofibroma, as in the
present case, also known as elephantiasis neurofibromatosa
has an overgrowth of epidermal and subcutaneous tissue
associated with a wrinkled and pendulous appearance, poorly
circumscribed, infiltrate along connective tissue septae, and
envelop normal structures with uniform fibrillary collagen
bundles [26]. There is an increased risk of malignant
transformation (from 12 to 29%) in these neurofibromas, that
necessitate follow-up clinically and with serial imaging with
tumor excision if the mass increases suddenly. Other
complications include bleeding from trauma, neurological,
limb movement, and psychological problems.
Surgical excision of cutaneous neurofibromas is the only
treatment option for most of the lesions in NF1 and is indicated
if they are causing discomfort or are visible and stigmatizing or
compromise organ function. Total excision of a plexiform neurofibroma is difficult due to finger-like fronds that insinuate
themselves into adjacent tissues which explain the local
recurrence in the present case, and so clinical follow-up is
required every 6 months during the first 2 years and then
annually. They are often vascular causing substantial blood loss
during removal and so careful imaging of tumors is essential to
define its boundaries and evaluate its vascular supply [27, 28]. Plexiform neurofibroma is not sensitive to chemotherapy and radiotherapy should be limited to malignant tumors because it
can stimulate the growth of plexiform lesions.
Conclusion
In conclusion, despite its rarity, physicians should consider neurofibroma as a differential diagnosis of a vulval swelling, especially in patients who have other clinical manifestations of neurofibromatosis, or with family history of the disease, that might help in the management by wide excision of the mass with a safety margin at the primary surgery to prevent its recurrence.
References
- Sengupta SP. Tumours and cysts. In: Long and Short cases in Surgery. 1st Edition, New Centre Book Agency Publications, Calcutta. 1996; 39-75.
- Diehl SR, Boehnke M, Erickson RP, Ploughman LM, Seiler KA, et al. A refined genetic map of the region of chromosome 17 surrounding the von Recklinghausen neurofibromatosis (NFI) gene. Am J Hum Genet. 1989; 44(1): 33-37.
- DʼAgostina AN, Soule EH, Miller RH. Sarcomas of the peripheral nerves and somatic soft tissues associated with multiple neurofibromatosis (von Recklinghausenʼs disease). Cancer.1963; 16: 1015-1027.
- Patil S, Chamberlain RS. Neoplasms Associated with Germline and Somatic NF1 Gene Mutations. Oncologist. 2012; 17: 101-116. doi: 10.1634/ theoncologist.2010-0181
- Ruggieri M, Huson SM. The clinical and diagnostic implications of mosaicism in the neurofibromatoses. Neurology. 2001; 56(11): 1433-1443.
- Wozniak W, Karwacki MW. Is “watchful waiting” superior to surgery in children with neurofibromatosis type 1 presenting with extracranial and extramedullary tumor mass at diagnosis?. Childs Nerv Syst. 2008; 24(12): 1431-1436. doi: 10.1007/s00381-008-0668-7
- Pascual CI, Lopez PP, Savasta S, Lopez SC, Lago CM, et al. Neurofibromatosis type 1 with external genitalia involvement presentation of 4 patients. J Pediatr Surg. 2008; 43(11): 1998-2003. doi: 10.1016/j.jpedsurg.2008.01.074
- Dogra BB, Ahmed S, Kandari A, Virmani R. Plexiform neurofibromatosis of vulva. Int J Res Med Sci. 2014; 2(4): 1771-1773. doi: 10.5455/2320-6012. ijrms201411109
- Packer RJ, Rosser T. Therapy for plexiform neurofibromas in children with neurofibromatosis 1: an overview. J Child Neurol. 2002; 17(8): 638-41. doi: 10.1177/088307380201700816
- Blickstein I, Lurie S. The gynaecological problems of neurofibromatosis. Aust N Z J Obstet Gynaecol. 1990; 30(4): 380-382. doi: 10.1111/j.1479-828X.1990. tb02036.x
- Thomas WJ, Bevan HE, Hooper DG, Downey EJ. Malignant schwannoma of the clitoris in a 1-year–old child. Cancer. 1989; 63 (11):2216-9.
- Kaefer M, Adams MC, Rinck RC, Keating MA. Principles in management of complex pediatric genitourinary plexiform neurofibroma. Urology. 1997; 49(6): 936-40.
- Griebel ML, Redman JF, Kemp SF, Elders MJ. Hypertrophy of clitoral hood: presenting sign of neurofibromatosis in female child. Urology. 1991; 37(4): 337-9.
- Nasir AA, Abdur-Rahman LO, Ibrahim KO, Adegoke MA, Afolabi JK, and AdeniranJO. Genitourinary Plexiform Neurofibroma Mimicking Sacrococcygeal Teratoma. J Surg Tech Case Rep. 2012; 4(1): 50–52. doi: 10.4103/2006-8808.100356
- Weiss SW, Goldblum JR. Enzinger and Weissʼs Soft Tissue Tumors. 4th ed. St Louis, Mo: Mosby Inc; 2001
- Redlick FP, Shaw JC. Segmental neurofibromatosis follows blaschkoʼs lines or dermatomes depending on the cell line affected: Case report and literature review. J Cutan Med Surg. 2004; 8(5): 353-6. doi: 10.1177/120347540400800505
- Kathry N N. Clinical aspects of neurofibromatosis I, Nurth European Journal of Paediatric Neurology 1998; 2(5): 223-231
- Tsang E, Birch P, Friedman JM. Valuing gene testing in children with possible neurofibromatosis1. Clin Genet. 2012; 82(6): 591-3. doi: 10.1111/j.1399-0004.2011.01801.x
- Mulvihill JJ, Parry DM, Sherman JL, Pikus A, Kaiser-Kupfer MI, Eldridge R. Neurofibromatosis 1 (Recklinghausen disease) and neurofibromatosis 2 (bilateral acoustic neurofibromatosis): an update. Ann Intern Med. 1990; 113(1): 39-52.
- Carey JC, Laud JM, Hall BD. Penetrance and variability of neurofibromatosis: a genetic study of 60 families. Birth Defects Orig. Artic Ser.1979; 15(5B): 271-281.
- Tonsgard JH, Yelavarthi KK, Cushner S, Short MP, Lindgren V. Do NF1 gene deletions result in a characteristic phenotype? Am J Med Genet. 1997; 73(1): 80-86. doi: 10.1002/(SICI)1096-8628(19971128)73:1
- Friedman JM, Arbiser J, Epstein JA, Gutmann DH, Huot SJ, Lin AE et al. Cardiovascular disease in neurofibromatosis 1: report of the NF1 cardiovascular task force. Genet Med. 2002; 4 (3): 105-11.
- Joshua TD, Janice MK, Jonathan WW, Gibbs R A, Borch R F, Tainsky MA et al. Molecular targets for emerging anti-tumor therapies for neurofibromatosis type 1. biochemical pharmacology. 2006; 72: 1485-1492.
- Sorensen SA, Mulvihill JJ, Nielsen A. Long-term follow-up of von Recklinghausen neurofibromatosis. Survival and malignant neoplasms. N Engl J med. 1986; 314(16): 1010-1015. doi: 10.1056/NEJM198604173141603
- Pivnick EK, Riccardi VM. The Neurofibromatoses. In: Freedberg IM, Eisen AZ,Wolff K, Austen KF, Goldsmith LA, Kartz SI, Fitzpatrick TB. Fitzpatrickʼs Dermatology in General Medicine. New York: Mc graw Hill. 1999; 2152-58.
- Harper JI. Genetics and genodermatoses. In: Champion RH, Burton JL, Burns DA, Breathnach SM editors Rook/Wilkinson/Ebling. Text book of Dermatology. 6th ed. Oxford: Blackwell Science. 1998; 378-84.
- Littlewood AH, Stilwell JH. The vascular features of plexiform neurofibroma with some observations on the importance of pre-operative angiography and the value of pre-operative intra-arterial embolization. Br J Plast Surg. 1983; 36(4): 501-506.
- Tonsgard JH, Short MP, Yamini B, et al: Surgical treatment of neurofibromatosis, in Schmidek HH, Roberts DW(eds): Schmidek and Sweetʼs Operative Neurosurgical Techniques, 5th Edition. Amsterdam 2006; 1: 968-974.
