

Research Article
The Biologic Nature and Function of Cancer Vaccines
1Division of Surgical Oncology, Department of Pathology, North Shore University Hospital, Northwell Health system, Hosfstra College of Medicine, Manhasset NY, USA
2Precision Biologics, Great Neck, NY 11021, United States
*Corresponding author: Myron Arlen, Department of Surgery, North Shore University Hospital, Northwell Health system, Hofstra College of Medicine and Precision Biologics, USA, E-mail: myronarlen@yahoo.com
Received: May 27, 2017 Accepted: July 10, 2017 Published: July 17, 2017
Citation: Arlen M, Arlen P, Tsang K, et al. The Biologic Nature and Function of Cancer Vaccines. Madridge J Vaccines. 2017; 1(1): 27-33. doi: 10.18689/mjv-1000107
Copyright: © 2017 The Author(s). This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Vaccines; Metastasis; Antibodies; Cystic fibrosis.
Introduction
Vaccines are biologic products that were developed to protect the host against foreign invaders such as bacteria and viruses thru enhancement of their innate host immune system. There is frequently a great deal of cross reactivity targeting unwanted sites when commercial immunogens (vaccines) are employed for a wide range of therapeutic approaches. True specificity is lost when using these vaccines to attack an array of invaders among which the cancer cell and associated conditions may be included. Here normal cells are targeted since similar surface antigens may be expressed and the mechanism of cellular destruction of any existing cancer cell may be compromised.
When tumor membrane antigens, no matter how small in number they may be expressed, are incorporated into a vaccine whose composition is typified by the addition of proteins such the epidermal growth factors and vascular growth factors, cross reactivity commonly occurs with adjacent normal tissue. This is a contradiction to the needed vaccine function.
In the case where one develops a specific tumor vaccine, the important component is that of the immunogenic protein which characterizes the tumor. It should contain no component reacting with other tumors of a different class nor with normal cells. The true tumor immunogen used in the development of a cancer vaccine thus has a different source than just tumor membrane, and is frequently derived from an oncofetal protein appearing in the fetus.
Oncofetal antigens are proteins for the most part [1]. They are typically expressed only during fetal development after which their genes are remethylated to eliminate their presence and thus loss of functin. They may be found as a modified molecule in adult cells with various associated malignancies (the tumorantigen) having evolved during the post fetal period by a virus or carcinogen demethylating the protein [2] and inducing a post translational modification of the initial onco fetal protein. These oncogenic proteins are often measurable in the blood of individuals with cancer and may be used to both diagnose and treat the tumors. The latter can occur when a thresh hold level of antigen is delivered. One example of an oncofetal antigen is alphafetoprotein, which is produced by hepatocellular carcinoma and some germ cell tumors. Another example is carcinoembryonic antigen, which is elevated in people with colon cancer and other tumors [3]. Other oncofetal antigens are the trophoblast glycoprotein precursor] and immature laminin receptor protein (also known as oncofetal antigen protein). Oncofetal antigens as they are being identified, act as promising targets for vaccination and have been used clinically against several types of cancers in a crude form derived from pooled allogeneic antigen [4].
Unfortunately the mutated oncogenic component found to eventually reside in the early transforming cancer cell is present at levels too low to be defined by the hosts immune system and as such the developing cancer is never routinely rejected. An early developing inflammatory process on the other hand, expreses that level of antigen in the infectious state that is easily recognized by the host. Here the form of disease is spontaneously brought under control thru an effective immune response, never seen with the onset of a developing malignancy.
Additionally the truer form of a cancer antigen, that is of oncofetal origin never mutates as the other so called tumor antigens do. The mutated form of the oncofetal protein is almost always that of the type seen in the cancer cell It requires, as a means of recognition, a separate and distinct monoclonal antibody to easily recognize the malignant genotype as well as premalignant cells by IHC. The latter has now been shown to comprise a portion of a field effect that may act as a source of new tumor initiation. This is true when such a field has a number of genetic foci induced either by viral or carcinogenic transformation.
The major mechanism of tumor control by the cancer vaccine is via ADCC (antibody dependent cell cyttoxicity) and does not depend on cytotoxic T cells for most of its activity. We will describe the mechanisms by which the true cancer vaccine functions to destroy an existing lesion however previous attempts at using existing vaccines in therapeutic trials have suggested some activity arising in the cytotoxic group of immunocytes [5]. While the infectious disease vaccines generate their immune response via the cytotoxic T cell interaction, just the reverse occurs in the immune control of tumor. The antibody response to a good tumor vaccine in the form of an IgG1 that induces ADCC can persist for more than 20 years. At initiation the tumor molecule seen in the transforming cell remains unchanged maintining its mutated post translational modified oncofetal protein structure. This persists as a definitive target in the fully malignant lesion as a specific tumor marker. It continues to remain within each of the tumor cells without mutation even in developing metastatic lesions. In its suboptimal threshold level it can still be the target for specific vaccine activity. When delivered in a post surgical situation, within 3-4 months antibodies targeting the tumor begin to appear and will keep the reappearance of tumor under control for many years.
Discussion
The derivation of the true cancer vaccine is different than the majority of the biologic products that have been introduced for cancer management and resulting mechanisms of action are different. As such, few if any products have been introduced for clinical use that can be attributed to produce a true host immune response against tumor immunogens that one expects to see.
As a example, a fully human monoclonal antibody against epidermal growth factor was introduced but produced unwanted toxicity against skin and GI mucosa requiring it to be discontinued. Such cross reactivity to normal cells should not prevail in a cancer vaccine product
The Cancer Vaccine, as a biologic reagent is clinically used to control and or prevent the growth of an existing or progressive malignancy [6]. While immunologic approaches have made progress in treating the lymphomas and leukemias, little improvement has been seen with the immunotherapy of advanced solid tumors. Those lesions, the adenocarcinomas of colon, pancreas, lung, and ovarian disease have failed to effectively respond to any of the newer immunotherapeutic agents including the Check Point inhibitors . These treatments work by targeting molecules that serve as checks and balances on immune cells, preventing those cytotoxic T cells from actively participating in the process of tumor cell control. It may prove that later on in the clinical trials at Precision Biologics that an anti PD-1 agent will be added to our existing antitumor agents, to evaluate possible an improved therapeutic antibody response For the most part the newer variations of vaccines now being developed represent an immunogenic post translational modification of an oncofetal protein that characterizes that malignancy. The modified protein is found in the malignancy only and demonstrates no cross reactivity to normal tissue. The reason for tumor growth without immune/host recognition is that the level of expression of the immunogen is far below the needed level for host recognition. When activated, its main function is that of producing a humoral response with the resultant main IgG1 targeting the tumor protein by ADCC (antibody dependent cell cytotoxicity) [7]. Of more importance is that the antigen is expressed in the premalignant phase with its stability persisting and characterized thru the transforming phase of the tumorgrowth (Fig.1).
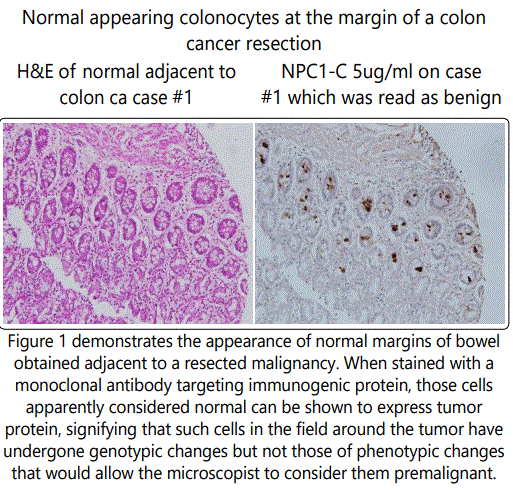
If we are treating metastasis from a previously resected primary, going back to the original primary provides information on the proper antigen being expressed in the metastatic lesion and as such the one needed to target the new site and treat the metastasis with the corresponding necessary reagent. The antibody developed against the oncofetal protein such as MUC5ac which predominates in the developing fetal bowel will not react with the malignant post translational modified antigen seen in colon cancer. Similarly that antibody Neo 102 targeting mutated MUC5ac found in the malignant lesion will not cross react with fetal MUC5ac protein which if not terminated in production at the time of fetal delivery, will lead to cystic fibrosis. When examining those monoclonals currently being employed to address a developing tumor, the mAbs against the epidermal growth factors, those against the vascular endothelial growth factors and those targeting the carbohydrate antigens such as Ca19.9 and Ca 17.1a are noted to be expressed in normal tissue during an inflammatory process and at various periods of time such targets mutate so that the interaction with specific mAbs and the target protein becomes non functional. One must decide whether sequencing the mutated protein and re: designing a functional mAb will in the long run equivocate with defining that specific immunogenic protein derived from the tumor oncofetal protein that can be defined at the initiation of the oncogenic process and which remains unchanged thru the natural history of that malignancy.
The first Cancer Vaccine to be employed for the therapy of malignant lesions was developed at Sloan Kettering by Bradley Coley. He noted that in many instances, the development of inflammation in the vicinity of certain tumors such as sarcomas and osteogenic sarcoma resulted in tumor regression. The result, Coleys toxin, a bacterial toxin derived from surrounding micro organisms was found to be effective in treating large primary and metastatic sarcomas. The first concept of this type of cancer vaccine was based on Coleys noting that when an erysipelas infection occurred near or in a sarcoma, regression of tumor was occassionally noted. This led Coley at Sloan Kettering to develop his vaccine comprised of strep pyogenes and staph seratia. Many sacomatous lesions were noted to regress [8].
Alexander Brunschweig, Chief of the Gyn Service initiated the concept of immune control of tumor. He recruited Chester Southam to help resolve certain issues. Southam, the first immunologist at Sloan-Ketttering, began initially working with HeLa cells in patients with various malignancies. He used local resection for the occasional transplant that illustrated a pattern of growth at the injection site.
Southam initiated a study with inmates at the Ohio State Prison System, delivering B16 melanoma cells to healthy volunteers. Rejection was noted within 20 days, but by repeating the same injection to the same volunteers one or two months later, rejection was noted in about 10 days defining First and Second set immune reactions.
Shortly after this he began testing B16 melanoma cells in patients with cancer at Sloan Kettering. This now resulted in rapid metastasis of the melanoma. There had been no previous experience in seeing how a tumor transplant evolved in patients with active malignant disease.
In the Keynote speech delivered at the International Vaccine 2017 meeting in Baltimore Md., it was apparent that the origin of the Immunogen and nature of the structure and function of Cancer Vaccines was poorly understood. The initial notation that such vaccines serving to treat cancer were oncofetal in origin and expressed far below the needed threshold level to be recognized by the host, were poorly understood.
This basic concept of tumor rejection or failure to eliminate a developing malignancy was based on comments by Richmond Prehn in the 1950's, that cancer was a foreign cell within the host, not too different from bacteria and or viruses that had invaded. His concept was that a cell such as the bacterial cell would be rejected because there was a threshold level of such needed antigen recognized by the hosts immune system. In the case of tumor cells, there was an immunogenic protein characterizing the tumor that was present, but far below that threshold level needed for cellular rejection. It was not recognized at the time as to whether the immune response to the invader was a humoral or cellular one. Most suspected the cytotoxic T cells as the major host response initiated against removing a growing neoplasm.
Based on studies by Rosenberg when examining tumor infiltrating lymphocytes (TIL) it was felt that the invading WBC represented a cytotoxic CD8 cell invading tumor to bring it under control. This did not explain why the immune response did not become initiated at inception of the tumor growth. It was noted that the TIL (tumor infiltrating lymphocytes) [9], when scaled up did not result in regression of tumor nor did the introduction of LAK (Lymphokine Actived Killer ) cells offer any further enhancement in tumor control. In contrast one of the first studies we reported on with regard to tumor invasion by immunocytes was that of the effects of such infiltration in patients with medullary cancer of the breast. Here it was noted that the greater the degree of lymphocyte involvement the poorer the prognosis Failure to reject early onset cancer suggested to Prehn [10], that the initial tumor growth was not recognized by the host immune system. His interpretation was that while bacteria and viruses were growing in the host, such corresponding cells could be recognized due to their expression of a threshold level of specific cellular antigen. In terms of the cancer cell, Prehn felt that the levels of antigen expressed by the cancer cell were far below what was immunologically needed to initiate an immune response. He therefore carried out several basic experiments to prove his concepts. In the first study, mice were immunized with a carcinogen and an additional group, a separate virus resulting in two strains of tumor that grew in his mice system. When he grew a large adenocarcinoma, removed the whole tumor and extracted protein representing the tumor immunogen, a vaccine so produced did not protect animals from tumor challenge. When he pooled several lesions and then challenged them with the carcinoma the pooled material was effective in allowing tumor to grow. Challenging the protected immunized mice with viral tumor allowed the latter to grow. The conclusion he reached was that a vaccine to prevent tumor could be produced when the proper threshold level of antigen was given. In addition the vaccine had to be specific to the system being treated.
As noted, the various approaches being employed to define the specific tumor immunogen has been ineffective probably due to the inability to obtain and as such define antigenic material at sub diagnostic levels. Without use of pooled allogeneic membrane protein, the opportunities to choose the right antigen for further study is extremely difficult.
P. Arlen, following thru on developmental procedures at Precision Biologics described the methods that have been incorporated into the groups approach in cancer vaccine development as introduced by Hollinshead in the development of the first human cancer vaccines from pooled allogeneic operative tumor specimens. Those Immunogenic Neoantigens that were potentially evaluated for incorporation into a human cancer vaccine were derived from a membrane preparation of pooled allogeneic cancer cells derived from patients undergoing surgery. Membrane fractions were isolated and tested for immunogenicity and utilized in a clinical trial in patients with chemotherapy refractory metastatic cancer, i.e. colorectal Ca.. A positive correlation was observed in patients who were able to mount and sustain IgG1 response to the vaccine. Antibodies were developed and screened against isolated tumor antigen. Antigens so derived were then tested for sensitivity, specificity, and anti-tumor function. Neoantigens were identified in colon cancer with these functional molecules and shown to be derived from oncofetal antigens functioning within the fetus. Here, several years later after the genes had long been remethylated into developing adult tissue, the oncofetal gene becomes reactivated, producing in many instances, a post translational modification of the initial oncofetal protein. This new molecule, in the malignant growth, represents a modification of the immunogenic protein characterizing the tumor but again in the sub threshold level so as not to be recognized by the hosts immune system. In the case of pancreas cancer the process occurs up to 20 years after transfection and within the DCIS state before invasive cancer is recognized.
In attempting to clarify the significance and validity of Prehn interpretation of the molecule serving as the tumor immunogenic protein, Hollinshead at GWU, evaluated the need to obtain this tumor protein in a similar fashion but from pooled allogeneic human cancer specimens Her goal was to get a positive delayed cutaneous hypersensitivity reaction (DHR) from antigen obtained from human tumor specimens. Large numbers of membrane material from operative (25-50) specimens were pooled to allow potential threshold levels of antigen to be reached [11]. Initially lung cancers, those of bowel origin and melanoma were separately pooled, sonicated and fractionated by MW over a Sephdex gel column. Those fractions collected off the column sequentially, allowed antigen to be partially separated by M.W. and each of the tubes collected off the gel column, tested for the desired DHR, represented by a 10mm wheel at minimum for each listed positive reaction.
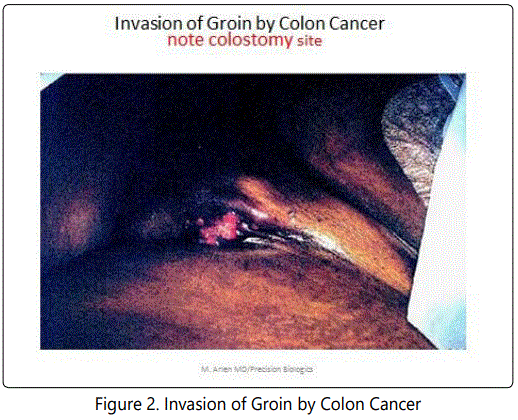
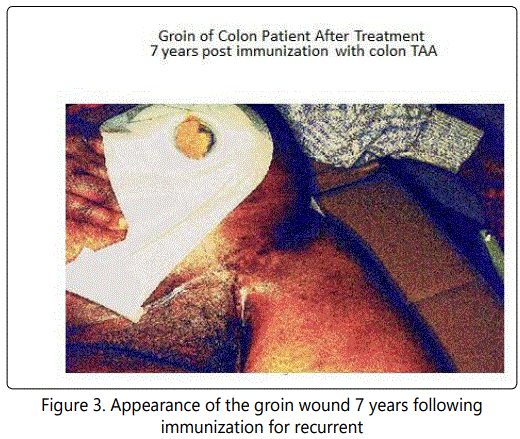
In period of time from the early 1970's to 80's there was little interference from the FDA and the hospital IRB in planning a potential vaccine project. If a product looked potientially viable and therapeutically non toxic then the patient could be approached to evaluate by placing them on a clinical trial and evaluating the immunogenicity of the tumor protein. When the Hollinshead antigen was provided to us via the surgical branch of the NCI to test for needed levels to induce a therapeutic response, within several weeks it was clearly shown that approximately 500-1000 ugms would be the effective dose. Examination of single solid tumor specimens indicated that few if any contained more than 20-50 ugms. Antigen, when employed as a vaccine, would be delivered in 3 divided doses along with an adjuvant. Prior to defining any mechanisms of action several patients were treated with interesting responses. In Fig 2 a patient had been referred with recurrent colon cancer, the lesion probable reappearing around the colostomy stoma following an APR. Tumor then grew down the adjacent left iliac vessels to enter the femoral triangle. Here it ulcerated out thru the skin With no specific approach available, vaccination was initiated. Over the weeks, regression of the groin lesion was noted and within months no further lesion was seen. See Fig. 3 shows the patient seven years following the initial immunization.
With a number of similar cases of recurrent or advanced resectable tumor demonstrating similar responses, the clinical trial for colon and lung cancer progressed. Five to 7 years later after we clearly showed responses in difficult cases, we approached FDA for permission to develop commercial methods for vaccine development. This was rejected due to the appearance of potential cases of clinical hepatitis B&C, HPV and HIV that could possible contaminate an operative specimen unknown to both patient and tumor.
At this point the FDA was willing to continue the trials but with the use of recombinant vaccines that would guarantee their consistency as well as purity.. This process would evolve by first producing monoclonal antibodies to the tumor antigen. They expected that even though the antigen was purified by gel electlophoresis that there would be an array of resulting monoclonals due to several antigens that would have migrated to the single point on the gel. They suspected correctly that no matter how definitive a band was on gel electrophoresis that the pooling of antigen of similar charge would result in a solitary gel band containing a number of different antigens. This proved true; we did define 3 discrete bands on HPLC that characterized the components of the colon cancer immunogenic array. These antigens were further purified, their monoclonals chimerized in CHO and used for affinity to isolate the purere antigen components for sequencing. Three well defined colon cancer antigens were identified and found to represent post translational modifications of the oncofetal proteins for bowel, that is primarily MUC5ac. CEAcam5,6 and A33 [12]. In non of the instances did the antibodies to the primary antigen react with their post translational modifications which were highly immunogenic but again expressed at subthreshold levels later in post fetal development.
Methods
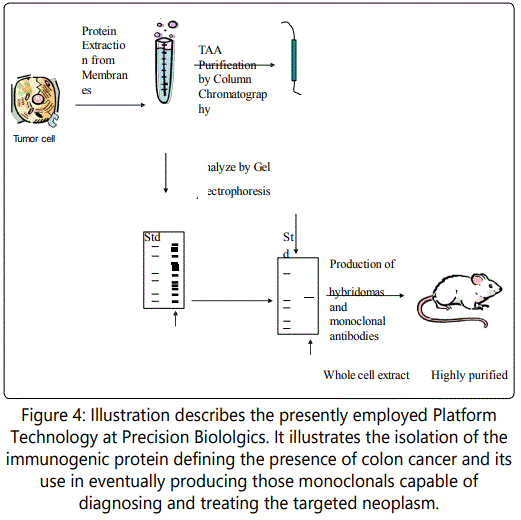
The platform technology that Precision Biologics has followed is illustrated in Fig.4 It outlines the basics from which we have derived our monoclonals and have moved on to tumor antigen identification. The later occurred by using affinity preparations for mass spectroscopy. With the antigen and associated monoclonals at hand we were able to show the effectiveness of the naked antibody in targeting nude mice challenged with human colon and pancreatic tumor transplants and to effectively demonstrate to the FDA the activity of the IgG1 as a primary mechanism in suppressing tumor growth.
Precision Biologics Platform Technology mAb's derived from Immunogenic T A A 's
Attempts at illustrating the role of Cytotoxic T cells in playing a major role in control of tumor growth could not be demonstrated.
This latter mechanism appeared reserved for eliminating populations of bacteria and viruses that had invaded the host. When the vaccine was delivered at its optimized level along with an adjuvant, high titers of a functional monoclonal were observed to be present in circulation for many years. Such patients remained free of disease during this period of time.
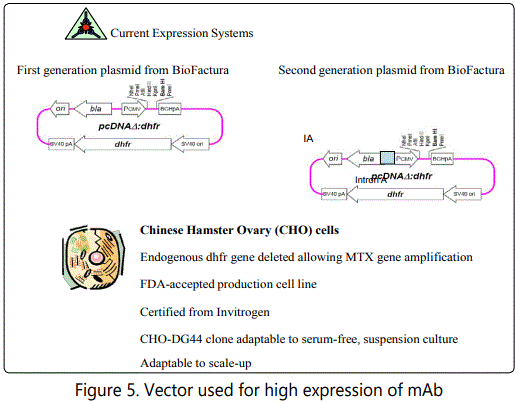
Preparations of monoclonal 101 were further upgraded. The output employed a high expression vector system to produce Neo-102. Our initial preparation of Neo 101 targeting those lesions expressing MUC5ac expressed approximately 140 mg. mAb per Litre of bioreactor fluid. From a commercial level this was felt to be too low. Changing the vector resulted in nearly 2000 mg/L. Here it was necessary to demonstrate Biosimilarity. When Neo-101 was compared to Neo102, not only was there a greater output per litre but antitumor activity was enhanced by at least 20% and the findings of slight hemolysis that was occasionally demonstrated with Neo101, totally eliminated. Since the molecular structure of the two molecules were identical it was felt that Neo-102 when chaperoned to the endoplasmic reticulum for folding was then passed on to the Golgi where a slight shift in its glycosylation pattern occurred. In Fig.5 we see how changes in the system employed can eventually lead to the therapeutic levels needed to come up with a product for eventual commercial use.
MUC5AC The mucins themselves are high molecular weight glycoproteins with O-linked oligosaccharides attached to serine or threonine residues of the apomucin protein backbone that is expressed in a cellular as well as tissue-specific pattern in normal tissues. This mucin family includes proteins that contain tandem repeat structures with a high proportion of prolines, threonines, and serines (which constitute the PTS domain) in the MUGastroenterology 6: 4. Additionally, MUC5AC in its modified form, is expressed ifn tumors of gastrointestinal, pancreatiobiloary, and endocervical origin (e.g., colon, esophagus, liver, lung, pancreas, stomach, and uterus). The study we performed, provided the peptidomimetics of an NPC-1 epitope derived from MUC5AC, including composition comprising the amino acid sequence of the epitope binding site defined by Phage display. Immunization of an animal with a fusion protein comprising a polypeptide of F(PHE) P(PRO) E(GLU) D(ASP) Y(TYR) F(PHE) R(ARG) Y(TYR) T(THR) N(ASN) Q((GLN) K(GLY) followed fusing a spleen cell with a myeloma cell, culturing post-fusion cells and hybridoma selection indicated that the peptide vaccines could function to induce a needed immune response. Prior to use in patients post surgical resection the various polypeptieds defined will be studied for optimum activity. i.e. FPEDYFRYTNQK (SEQ ID NO: 4) or SLPDDWFRYINY (SEQ ID NO: 5),
While it is apparent that in treating cancer patients such as those colon or pancreatic cancers that have metastasized and having failed all known therapeutic modalities have a life span limited to weeks, an infusion of antibody is required since an antitumor response can be seen in a matter of hrs. When immunization with a vaccine occurs it requires several months till the proper level of antiboidy results and is effective in controlling reappearance of tumor post surgery. In those patients originally treated with the Hollinshead vaccine, significant levels of mAb could be detected 20 or more years post immunization.
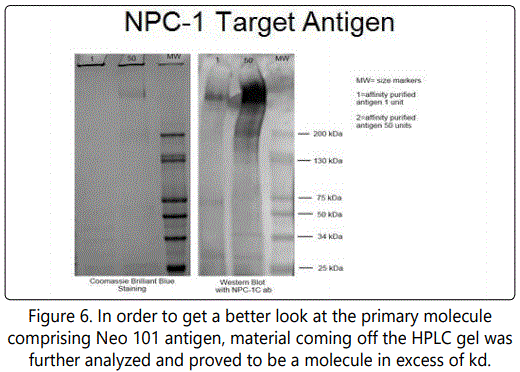
By removing said animal's spleen and preparing a single cell suspension, fusing a spleen cell with a myeloma cell, culturing post-fusion cells in hybridoma selection medium, culturing the resultant hybridomas, screening for specific antibody production, and selecting hybridomas which produce the desired antibody.copy result, Small polyps (SP), multiple polyps (MP), large polyps (LP), and colon cancer (CC) can be defined. The results suggests a correlation between the level of NPC-1 antigen detecting in a stool sample with the presence of polyps and/or colon cancer, where the higher the NPC-1 antigen amount detected is correlated the larger the polyps or the higher the number of polyps. Further, the data is suggestive of the high levels of NPC-1 antigen (e.g., over 20,000 units of NPC-1 antigen in a sample) as indicative of colon cancer.
The study we performed as part of the clinical trial defined the sequence of the mutated MUC5ac protein and provided the peptidomimetics of an NPC-1 epitope derived from MUC5ac. This included composition comprising the amino acid sequence of the epitope binding site defined by Phage display. That is the polypeptide: F(PHE) P(PRO) E(GLU) D(ASP) Y(TYR) F(PHE) R(ARG) Y(TYR) T(THN(ASN) Q((GLN) K(GLY). This sequence proved to be that fragment of the 600kd MUC5ac molecule responsible for antibody production and thus could potentially be utilized for eventual peptide vaccine therapy as a preventative following surgical resection in high risk patients.
In examining a higher powered view of the colon cancer (IHC) specimen defined in Fig 7,we can see the effect of doublestainiing which suggests that at a later time doble mAb may be more effective. , At 100 power it is apparent that those cells at the margin of the intestinal villi are expressing tumor protein which was expected to be shed into the lumen of the bowel. We have developed as a consequence of such observations a simple office stool ELISA test to exam the stool of a patient for diagnostic purposes. When we can show the presence of a small amount of antigen present in the stool, it would represent the presence of an early intraepithelial lesion unrelated to polyp developing within a field effect of the bowel. Absence of antigen would signify that the bowel is free of any developing malignant lesion and that colonoscopy can be delayed.
When examining the field around a colon cancer malignancy it has become apparent that the primary itself has arisen from surrounding colonocytes that appear normal. IHC of this tissue surrounding the neoplasm clearly shows that many cluster of normal appearing colonocytes are expressing antigen suggesting that such tumors do not arise from polyps for the most part but from transforming colonocytes that appear to express normal phenotypic features but have undergone genomic changes categorizing them as premalignant. Leaving such cells behind will result in anastomotic recurrence [13].
The data obtained from the initial monoclonal trial is leading to a planned for clinical Phase III study where low dose chemotherapy will be added to remove inhibitory blocking substances shown to be expressed by the tumor Eventually additional therapeutic modalities will be added to the chemoimmunotherapy planned for trial. Here IL-15 will be considered to enhance ADCC and an eventual radiolabelled alpha emitter of Neo-102 will utilized to enhance the overall response.
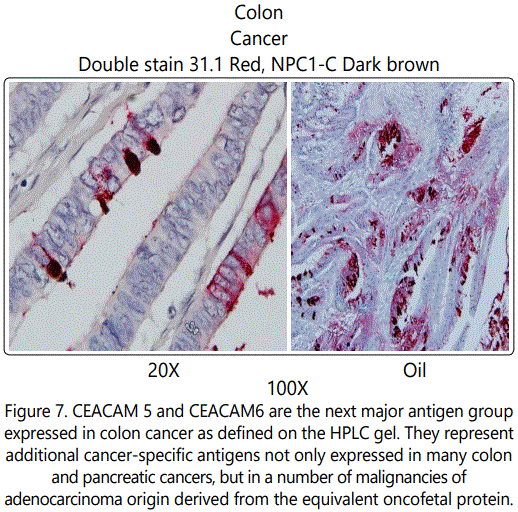
Figure 7. CEACAM 5 and CEACAM6 are the next major antigen group expressed in colon cancer as defined on the HPLC gel. They represent additional cancer-specific antigens not only expressed in many colon and pancreatic cancers, but in a number of malignancies of adenocarcinoma origin derived from the equivalent oncofetal protein. Using the antibody to target the antigen surprisingly shows high levels of expression in triple negative breast cancer which may indicate an additional approach than the present immunochemothrapy approach that has been employed. In contrast to the CEAs which are noted in many normal glandular tissues, these modified proteins are only found in the malignant phenotypes and have the capability of being detected in many malignant lesions, but have therapeutic efficacy thru strong ADCC. The carcinoembryonic antigen (CEA) gene family is a member of the IgCAM superfamily including 29 related genes and pseudogenes. These CEA proteins function as intercellular hemophilic and heterophilic adhesion molecules and do have additional signaling properties. Carcinoembryonic cell adhesion molecule (CEACAM) 5 and CEACAM6 share ˜90% homology in the N domain but differ in the number of IgC2-like domains (A and B domains). Both proteins contain a glycosylphosphatidylinositol (GPI) membrane anchor and are targeted to lipid rafts in apical membranes of polarized epithelial cells. CEACAM5 and CEACAM6 bind a variety of gram-negative bacteria and mediate internalization /phagocytosis, participating in innate immune defense in the intestine. CEACAM6 expression is elevated in many solid tumors, but variable as a function of histotype. Based on previous work demonstrating a role for CEACAM6 in tumor cell migration, invasion and adhesion, and formation of distant metastases, it may be a promising target for antibody-based therapy [14].
On HPLC fractionation of the Hollinshead preparation 3 distinct immunogenis comprised the pooled vaccine. The above 2 that were described were the MUC5ac and CEACAM5ac comprised the major antigenic positions. These proved to be oncofetal in origin. The 3rd molecule defined as A33 by Lloyd Old at Sloan Kettering was shown to be a common membrane protein but was Unable to induce a strong antitumor response when antibodies to the A33 molecule was delivered IV. When the post tranlation oncofeltal form was delivered, the ADCC effect was more than 10% active than mAbs Neo 102 and 201. As studies are completed with the last 2 GM: P mabs be studying A33 in vivo.
In 2015 at the ASCO GI Symposium in San Francisco, CA, results of the Phase 1 study of Ensituximab (Neo-102) in chemotherapy refractory metastatic colorectal Cancer Study were presented in a poster session. The study revealed a Maximal Tolerated Dose of 3.0 mg/kg IV every 2 weeks. The overall survival observed in this demonstrated 10.4 months comparing favorably to the historical control for a similar population of patients with advanced colorectal Ca (5 months). This led to a larger Phase 2 multi-center colorectal cancer study using Ensituximab in the same patient population where results are being confirmed. Because tumors do shed an inhibitory molecule into the serum blocking many of the effects of the anti-tumor IgG1 in its ADCC response, new trials are being designed to initiate the study somewhat earlier and with low dose chemo being added to minimize inhibitory antigen. This will follow with the eventual addition of IL-15 which can enhance ADCC thru further activation of the NK cell effect. When all factors are in place we anticipate that there will be a marked improvement in the management of patients with recurrent colon and other cancers.
References
Contact us for any additional information - contact@madridge.org
 Madridge Publishers is licensed under a Creative Commons Attribution 4.0 International License.
Madridge Publishers is licensed under a Creative Commons Attribution 4.0 International License.