
1School of Pharmacy, University of Reading, Reading, UK
2School of Pharmacy, University of Reading Malaysia, Malaysia
3Institute for Cardiovascular and Metabolic Research, University of Reading, UK
*Authors DR, MS and AA, and ♯HMIO and SV contributed equally to this work.
Background: Platelets (small circulating blood cells) play indispensable roles in the regulation of haemostasis via blood clotting. However, their inappropriate activation leads to thrombosis, which obstructs blood flow to major organs such as heart and brain resulting in heart attack and stroke respectively. Hence, platelets act as a promising target to treat/prevent cardiovascular diseases (primarily thrombotic diseases). Direct relationships between cardiovascular health and dietary flavonoids have been extensively studied for a long period. Nevertheless, numerous challenges are associated with the use of dietary components in biological systems for the prevention and treatment of diseases. Some of these include the poor absorption in intestine, the reduced bioavailability in blood stream, inability to readily cross the cell membranes and their modest stability in biological systems. Here, we report the design, synthesis, chemical characterisation and biological evaluation of Ruthenium complexes of chrysin(a natural flavonoid), and its synthesised thioflavone derivative for the modulation of platelet function and thrombus formation.
Methods and Results: In this study, we analysed the effects of chrysin in the modulation of platelet reactivity using washed human platelets and platelet-rich plasma by optical aggregometry. Similar to other flavonoids, chrysin displayed substantially reduced effectswhen platelet-rich plasma was used in comparison to washed platelets. In order to intensify the effects of chrysin under physiological conditions such as in whole blood, its sulphur (thio-chrysin) and Ruthenium (Ru-chrysin and Ru-thio-chrysin) conjugated synthetic derivatives were synthesised and characterised. The effects of synthetic chrysin derivatives were examined in platelets using a variety ofplatelet functional assays such as aggregation, the measurement of fibrinogen binding and P-selectin levels, calcium mobilisation and in vitro thrombus formation. In comparison to natural chrysin, Ruthenium- conjugated chrysin derivatives, specifically Ru-thio-chrysin significantly reduced distinctive functions of plateletsand thrombus formation under arterial flow conditions at minimum of 6.25µM. Together, these results demonstrate that Ruthenium- based synthetic chrysin derivatives exert enhanced inhibitory effects in platelets under physiological conditions.
Conclusions: This study highlights the importance of Ruthenium-conjugated synthetic flavonoid derivatives in the modulation of platelet function and thrombus formation. Due to their numerous beneficial effects in biological systems, Ruthenium-conjugated molecules will be greatlyvaluable in therapeutical applications for the prevention and treatment of cardiovascular (particularly thrombotic) diseases.
Biography:
Dr. Sakthivel Vaiyapuri is a Lecturer in Pharmacology in the School of Pharmacy at the University of Reading, UK. I completed my Bachelors in Biochemistry at Bharathidasan University and Masters in Biotechnology at the University of Madras, India. Ireceived my PhD in the field of snake venoms and postdoctoral experience in cardiovascular diseases with specific interest on platelet signalling from the University of Reading. Currently, my research group involved in the functional characterisation of inflammatory molecules such as formyl peptide and toll-like receptors in the modulation of platelet function at the interface between thrombosis and inflammation. With the better understanding of these receptors, we develop target-specific chemical molecules in collaboration with colleagues in Pharmaceutical chemistry and evaluate their biological effects in the modulation of thromboinflammatory responses. Furthermore, we are also engaged in analysing the toxic components of snake venoms and their impact on various functions of cardiovascular system.
1Ajman University of Science and Technology, UAE
2Temple University, Philadelphia, USA
Purpose: The goal of this study was to modify structurally our low affinity lead lactones in an effort to improve receptor affinity. The design of new chemical entities for muscarinic receptors was based on molecular modeling and structure activity relationship studies from the literature and our previous work.
Methods: Lead Lactone-containing compounds previously synthesized in our laboratory demonstrated moderate affinity for muscarinic receptors. Structure activity relationship (SAR) data from the literature suggest that specific amine-containing functionalities repeatedly appear in high affinity muscarinic ligands. Based on these reports, various lead modification approaches were utilized in order to design a series of novel muscarinic ligands containing the lactone ring (or bioisostere) and an aminecontaining fragment reported in other high affinity ligands. Homologation approach was selected in the design of a series of lactone based compounds. Bioisosteric approach for the lactone ring replacement (e.g., substituted tetrahydrofuran, 1,3-benzodioxoles, oxazolidinones and chromone) were also investigated. The need for rapid and efficient routes to the scaffolds prompted the development of several highly efficient synthetic methods to structurally diverse target compounds. Test compounds were evaluated for affinity in muscarinic binding assays. The ligands exhibiting % specific inhibition >50% were selected for further evaluation (IC50 data and ultimately subtype selectivity).
Results: A lactone-based ligand having diphenylmethylpiperazine fragment was identified as non-selective muscarinic ligand with IC50 of 340 nM whereas its homolog was found to have higher affinity (IC50 of 17 nM). Preliminary binding study suggests that substituted tetrahydrofuran, 1,3-benzodioxoles, oxazolidinones and chromone nuclei represent possible bioisosteric replacements for the lactone ring in the series of ligands evaluated herein. The ligands evaluated for subtype selectivity in the muscarinic assays were found to be non-selective.
Conclusions: Novel synthetic routes were developed for the synthesis of target compounds. The compounds reported herein represent an interesting series of novel muscarinic ligands that require further study. The design of future ligands will be based on the SAR data supplied in this work as well as SAR studies reported in the literature.
Biography:
Richie R. Bhandare received his B.Pharm and M.Pharm from University of Mumbai and PhD in Medicinal Chemistry from School of Pharmacy, Temple University, Philadelphia, USA in 2013. He was working at a leading Pharmacy College in Mumbai as an Assistant Professor in Pharmaceutical Chemistry. He has guided 13 M.Pharm students. He works in the area of designing novel compounds for muscarinic, serotonergic and retinoic receptors and organic reaction method development. He will now be associated with College of Pharmacy and Health Sciences, Ajman University of Science and Technology, Ajman, UAE as an Assistant Professor in Medicinal Chemistry.
Novosibirsk State University, Russia
Nanocomposites consisting of titanium dioxide nanoparticles (TiO2)and oligonucleotides or their analogs were prepared to deliver nucleic acid-based compounds into cells. The nanocomposites (TiO2•PL-oligo)were designed by the immobilization of polylysine-containing oligonucleotideson TiO2 nanoparticles. It was shown that the proposed nanocomposites exhibited a low toxicity and very high activity against influenza A virus (IAV) in vitro and in vivo. The nanocomposites bearing the oligonucleotide targeted to the chosen regions of (-)RNA and (+)RNA of segment 5 of different IAV subtypes inhibited the virus reproduction by >99%. Moreover, it was shown a possibility of using the proposed nanocomposites for the treatment of hypertensive disease by introducing them into the hypertensive ISIAH ratsdeveloped as a model of the stress-sensitive arterial hypertension. Oligonucleotides should be targeted to some genes involved in the pathogenesis of essential hypertension. The angiotensin-converting enzyme (ACE) involved in the synthesis of angiotensin-II was chosen as a target. Two ways of administration (intraperitoneal injection and inhalation) were examined. Both methods showed a significant (by 20-30 mm/Hg) decrease in systolic blood pressure, when the nanocomposite contained the ACE gene-targeted oligonucleotide. When using the oligonucleotide with a random sequence, no effect was observed. Thus, it was demonstrated a possibility of using the proposed nanocomposites as efficient drugs to achieve a high biological efficacy.
“The work was supported by the grant of Russian Scientific Foundation no. 16-15-10073.”
Biography:
Asya S. Levina was born in Novosibirsk, Russia. She received the MS degree in chemistry from Novosibirsk State University, Russia, in 1969 and the Ph.D. degree in bioorganic chemistry from Novosibirsk Institute of Organic Chemistry in 1974. From 1969 to 1993, she worked in the Laboratory of Nucleic Acids Chemistry at the Institute of Bioorganic Chemistry. From 1993 to 1997, she was Senior Research Associate with the Laboratory in Worcester Foundation for Biomedical Research, Shrewsbury, MA, USA. Since 1997, she has been a Senior Research Scientist at the Institute of Chemical Biology and Fundamental Medicine, Siberian Branch of Russian Academy of Sciences. She is the author of more than 100 articles and more than 10 patents. Her research interests include the synthesis of oligonucleotides and their derivatives, preparation of DNA arrays, and the synthesis of nucleic acid-based nanocomposites. Currently she works at the Novosibirsk State University.
Ruhr University of Bochum, Faculty of Chemistry and Biochemistry, Germany
Bisphenols (BPs) are abundant in modern life, since they are widely used in numerous plastic products as plasticising agents. Humans are mainly exposed through canned foods to bisphenols, leading to the occurrence of BPs in human blood serum, urine, and sweat. Chemically, this family of compounds is characterised by an optionally substituted central sp3 carbon atom that is flanked by two hydroxyphenylfunctionalities. The protein K-Ras belongs to the family of small GTPases, which are enzymes that hydrolyse guanosine triphosphate (GTP) to guanosine diphosphate (GDP). Through switching between an active (GTP-loaded) and an inactive (GDP-loaded) state K-Ras acts as a molecular switch within cells. Furthermore, K-Ras is a known protein oncogene, because it is found to carry mutations in approx. 20 % of human cancers. Both, the activation states of K-Ras and the binding of small molecules to this GTPase can be investigated by nuclear magnetic resonance (NMR) spectroscopy. In addition, 1H-15N two-dimensional NMR spectra not only provide an opportunity to map the binding site of a ligand but also allow for the determination of the dissociation constant of a complex, a valuable parameter that helps to evaluate the impact of a ligand on a protein. In this work we show, based on our previous studies, that not only BPA but also analogues bind to K-Ras and we also discuss potential consequences of these interactions.
Biography:
Raphael Stoll is Professor of Biomolecular Spectroscopy in the Faculty of Chemistry and Biochemistry at the Ruhr University of Bochum, Germany. He studied Physiological Chemistry and Biochemistry at the Universities of Tübingen, Germany, and Oxford, UK, as a fellow of the „Studienstiftung” and DAAD. Supported by a fellowship from the FCI, he carried out his doctoral research at the Max Planck Institute for Biochemistry in Munich and received his PhD from the Technical University of Munich. After a stay as a research associate at The Scripps Research Institute, CA, USA funded by first a DAAD- and then an Emmy-Noether fellowship, he initially joined the Ruhr-University of Bochum as Junior professor. His research focuses in the main on structure-function-relationships of medically-relevant proteins, specifically those involved in the development of cancerous tumours, in order to better understand the causes of the condition and propose more effective treatment strategies.
Laboratoire Chimie et Biochimie de Molécules Bioactives – Université de Strasbourg/CNRS, France
Today, almost all important microbial infections throughout the world, such as tuberculosis, malaria, nosocomial diseases, are becoming resistant to antibiotics. Antimicrobial multi-drug resistance has been called one of the worldʼs most pressing public health problems. It is therefore urgent to find innovative targets for new antimicrobial drugs. Proteins involved in isoprenoid biosynthesis represent such targets. Isoprenoids are found in all living organisms and are essential for all bacteria. The alternative mevalonate-independent methylerythritol phosphate (MEP) pathway for the biosynthesis of isoprenoids, which is present in many pathogenic bacteria e.g. Mycobacterium tuberculosis, M. leprae, as well as in opportunistic pathogens e.g. enterobacteria, Acinetobacter spp., Pseudomonas spp., and present in the parasitic Plasmodium species responsible for malaria but absent in human represents an attractive target for the design and development of new antimicrobials. We focus on developing novel inhibitors for 1-deoxy-D-xylulose 5-phosphate reductoisomerase (DXR), the second enzyme of the MEP pathway for isoprenoid biosynthesis in pathogens.
Biography:
Dr. Catherine Grosdemange-Billiard is Professor in Chemistry at “Université de Strasbourg”, France. Her scientific background is in fields as varied as biochemistry, organic synthesis, surface chemistry, bioorganic chemistry and medicinal chemistry. After a B Sc. in Biochemistry, a M Sc. in Chemistry, a PhDin Physical Chemistry, she joined the Georges Whitesidesʼ group at Harvard University as a postdoctoral fellow. Back to France, she was appointed to assistant professor. In 1995, she moved to Michel Rohmerʼs group where she conducted research in chemistry and biochemistry of bacterial hopanoids. Since January 2014, she is the leader of the laboratory of “Chemistry and Biochemistry of Bioactive Molecules”. Her research interests are focused on the design of new antimicrobials.
University of Zululand, Republic of South Africa
The current rapid increase in incidences of cardiovascular events indicates a need for discovery of new more effective cardioprotective agents. This study evaluated the cardioprotective effect of methyl-3β-hydroxylanosta-9, 24-dien-21-oate (RA-3) from Protorhuslongifolia stem bark. The cardioprotective effect of RA-3 was investigated in isoproterenol-induced myocardial injury in high fat diet (HFD)fed rats. Rats were randomly divided into the normal diet (ND) fed group and high fat diet (HFD) fed groups. The experimental hyperlipidemicgroup was orally administered with RA-3 (100 mg/kg body weight) for 15 days. The rats were then injected with isoproterenol (85mg/kg b.w) to induce myocardial injury. At the end of the experimental period, hearts and blood samples were collected and used for histology and biochemical assays, respectively. RA-3 exhibited cardioprotective effect as it minimized myocardial injury in HFD fed rats. Few lesions of acute hyaline degeneration and reduced fat deposition were observed in the heart tissue of the triterpene-treated rats. Lactate dehydrogenase activity waseffectively decreased in the blood of the triterpene-treated rats(44.1mU/mL) compared to the untreated group (64.8mU/mL). The RA-3 treatment also significantly decreased levels of serum total cholesterol (7.51 mmol/L) and LDL-c (4.46 mmol/L) with an increase in HDL-c (47.3 mmol/L) in HFDinduced hyperlipidemic rats relative to untreated control group. An increased glutathione content and catalase activity along with lower levels of malondialdehyde in the triterpene-treated animals (120.8 nmole/µL) than in the non-treated HFD fed rats (143.6 nmole/µL) were also observed. The results indicate that the triterpene has cardioprotective effect. It is apparent the triterpene has a potential to be used in the prevention and treatment of cardiovascular diseases and related health problems.
Biography:
Dr. Rebamang Anthony Mosa has completed his PhD at the age of 35 years from University of Zululand. He is a senior lecturer at the University of Zululand and a registered member of the South African Council for Natural Scientific Professions (SACNASP). He has published over 15 papers in reputed journals. His area of expertise is in phytomedicine, currently focusing on bioactivities of plant-derived compounds against human metabolic disorders.
Aligarh Muslim University, India
In our study, renal cancer is induced in rats making use of dimethylnitrosamine (DMN). G1 – Group 1 were control rats and G2 – Group 2 rats were given a single intra-peritoneal injection of DMN of 50 mg/kg body weight resulting in 100% incidences of renal tumors after 12 months. SEM and histopathology confirmed the presence of renal cancer in the DMN-treated rats. Making use of ammonium sulfate precipitation and gel filtration chromatography on Sephacryl.
S-100HR column, a thiol protease inhibitor was isolated from kidney of control rats known as Rat kidney Cystatin (RKC) as well as from kidney of cancerous rat called as Cancerous Rat Kidney Cystatin (CRKC). Both these inhibitors were characterized, and interestingly, it was found that CRKC showed greater anti-papain activity and also it was stable in a broad pH and temperature range thus implying that CRKC is more stable as compared to RKC. UV and fluorescence spectroscopy point out in structural difference between RKC and CRKC which was further confirmed by Circular dichroism (CD) and FTIR spectroscopy. Our study clearly showed that kidney cystatin is structurally modified in the case of renal cancer and performs its role in a more efficacious manner.
Biography:
Mohd Anas Shamsi is a Research Fellow at the Department of Biochemistry, Aligarh Muslim University under the supervision of Prof. Bilqees Bano since 2014. He has published 8 papers in reputed journals of protein biochemistry and his work is primarily devoted to kidney mainly rat and buffalo kidney as these organisms are being used as model organisms for various studies related to humans. He completed his Bachelors with an aggregate of 81% and his Masters with an aggregate of 75%. He is currently a BSR-JRF Fellow and also has qualified CSIr-NET and GATE. He has attended several reputed confernce most recently being 4th Nanotoday held in Dubai, UAE.
1College of Medicine & Health Sciences, United Arab Emirates University, United Arab Emirates
2Institute of Pharmacy, University of Regensburg, Universitätsstraße 31, Germany
3Institute of Pharmacy and Food Chemistry, Julius-Maximilian University of Würzburg, Germany
Both the acetylcholine esterase (AChE) and the histamine H3 receptor (H3R) are involved in the metabolism and modulation of acetylcholine release and numerous other centrally acting neurotransmitters. Hence, dual-active AChE inhibitors (AChEIs) and H3R antagonists hold potential to treat cognitive disorders like Alzheimerʼs disease (AD). The novel dual-acting AChEI and H3R antagonistUW-MD-72 shows excellent selectivity profiles over the AChEʼsisoenzymebutyrylcholinesterase (BChE) as well as high and balanced in vitro affinities at both AChE and hH3R with IC50 of 5.4 µM on hAChEand hH3R antagonism with Ki of 2.54 µM, respectively. In the current study, the effects of UW-MD-72 (1.25, 2.5, and 5 mg/kg, i.p.) on memory deficits induced by scopolamine (SCO) and dizocilpine (DIZ) were investigatedin a step-through type passive avoidance paradigm in adult male rats applying donepezil (DOZ) and pitolisant (PIT) as reference drugs. The results show that acute systemic administration of UW-MD-72 significantly ameliorated the SCO- and DIZ-induced amnesic effects. Furthermore, the ameliorating activity of UW-MD-72 in DIZ-induced amnesia was partly reversed when rats were pretreated with zolantidine, but not with the H1R antagonist pyrilamine. Moreover, ameliorative effect of UW-MD-72 in DIZ-induced amnesia was strongly reversed when rats were pretreated with a combination of ZOL and SCO, indicating that these memory enhancing effects were partly observed through histaminergic H2R as well as muscarinic cholinergic neurotransmission. These results demonstrate the ameliorative effects of UW-MD-72 in two in-vivo memory models and provide evidence for the potential of dual-acting AChEI and H3R antagonists to treat cognitive disorders.
Biography:
Dr Bassem Sadek is Associate Professor of Pharmacology in College of Medicine and Health Sciences, UAEUniversity. He received his B. Pharm. in 1994 from The Free University of Berlin (FUB), Germany, and PhD in Medicinal Chemistry, Drug Design and Development in 1999 from FUB University (Germany). Over the years, he has developed innovative research projects which focused on the identification and manipulation of brain signaling regulating psychiatric as well as neurodegenerative disorders such as Alzheimer, addiction, depression, epilepsy, anxiety, and stress. Dr Sadek is a member of editorial board and a peer-reviewer to many international journals.
1Laboratory of Applied Chemistry & Environment, University Hassan 1er, Department of Chemistry, Morocco
2Laboratory of Biochemistry & Neuroscience, University Hassan 1er, Department of biology, Morocco
Many studies have reported that different parts of Opuntia genus (flower, skin fruit, seed, cladode) contains a high level of some bioactive antioxidant components such as phenolic acids, flavonoids, and vitamins, that have a lot of benefits at the term of nutritional, pharmacological, therapeutic, and health.
This work concerns the physicochemical analysis and evaluation of antioxidant capacity in cladode juices of two species of Opuntia from Settat-Casablanca region of Morocco. The objectif of this study was to determine the composition of phenolics, vitamin C and betalains, and the evaluation of antioxidant activity by 2, 2-diphenyl-1-picrylhydrazyl (DPPH) and the hydrogen peroxide scaneging (H2O2) in cladode juices.
The Total phenolics content are 1216,10 ± 0,03 and 1113,94 ± 0,02 mg of Gallic Acid Equivalent (GAE) /100 g of fresh weight, then both species indicate a similar quantities of vitamin C, 13,78 ± 0,04 mg of ascorbic acid /100 ml and 13,33 ± 0,05 of ascorbic acid /100 ml in Opuntia ficus indica and Opuntia megacantha respectively. The quantification of betalain pigments (betaxanthin and betacyanin) were higher in Opuntia ficus indica ( 5,12 ± 0,04 and 10,46 ±0,00 mg/100 g of fresh weight), than Opuntia megacantha one ( 3,87 ± 0,01 and 6,18 ± 0,00 mg/100 g of fresh weight). The species had comparable antioxidant activity in both, H2O2 and DPPH assays.
The analyzes of bioactive compounds from two species cladode juices had antioxidant activity and confirm their potential for exploiting the autochthonous of existing biodiversity of cactus, which makes this plant a wealth that should be better investigated and analyzed.
Keywords: Opuntia, juice cladodes, phenolics, vitamin C, betalains, antioxidants.
Albany College of Pharmacy and Health Sciences, USA
In the United States, fluoroquinolone antibiotics recently have gained increased attention due to safety concerns. In November 2015, a joint meeting of the Antimicrobial Drugs Advisory Committee and Drug Safety and Risk Management Advisory Committee was conducted to review risks and benefits of the systemic fluoroquinolone antibacterial drugs for the treatment of acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis in patients who have chronic obstructive pulmonary disease, and uncomplicated urinary tract infections. Potential fluoroquinolone-associated adverse events, both new and established, were a major focus of the Food and Drug Administration (FDA) advisory meeting discussion. Given emerging evidence that multiple toxicities can occur concurrently and result in potentially permanent disability, the condition “fluoroquinolone-associated disability,” or FQAD, was first described. In response to this advisory meeting, the FDA released a Drug Safety communication in May 2016 advising restricted use of fluoroquinolones for uncomplicated infections, as risks generally outweigh benefits. This presentation reviews established fluoroquinolone-associated adverse events, including tendinopathy, neurotoxicity, and arrhythmias, as well as emerging toxicities. Key findings from the FDA advisory meeting and FQAD data are also reviewed.
Biography:
Monique Bidell is currently an Assistant Professor of Pharmacy Practice at Albany College of Pharmacy and Health Sciences in Albany, NY. She maintains an active clinical practice site at St. Peterʼs Hospital in Albany, NY. Dr. Bidell earned her Doctor of Pharmacy (PharmD) degreefrom Northeastern University in Boston, MA. She completed a year of postdoctoral residency training in Pharmacy Practice at Beth Israel Deaconess Medical Center in Boston, MA. She then completed a specialty residency in Infectious Diseases at the University of Chicago Medicine in Chicago, IL. She is a Board Certified Pharmacotherapy Specialist.
Bharati Vidyapeethʼs College of Pharmacy, India
Cancer deaths rule the charts for over a decade with no persistent solution. Novel target specific therapy is an absolute need of the hour. Abundant work has been done and reported concerning the close liaison between inflammation and cancer. Curcumin is one such molecule that has been extensively explored against inflammation and is being recurrently studied against various types of cancer including leukemia with multiple mode of action, one amongst them being via histone methylation. However, the drawback of poor bioavailability profile restricts its drug status. Difluorinated chalcones and propanediones, owing to their structural similarity to the enol-keto form of curcumin, as well as to the plausibility of improved bioavailability due to fluorine atoms, seemed to bear appropriate candidature to be explored against histone methyltransferase in leukemia. These molecules were docked with 3K5K, G9a -UNC0224 crystal complex along with curcumin. Only after visible desired interactions, these molecules were subjected to enzyme inhibitory studies, considering BIX-01294 and curcumin positive control. 4-fluorine substituted di-fluorinated propanedione emerged as a potential inhibitor of histone methyltransferase highlighting 8-9% levels of methylation at 1 and 10 µM concentration. It showed significant apoptotic activity leading to G2/m arrest. In this study, we provide the first evidence that difluorinatedpropanedione is an inhibitor of histone methyltransferase and functionally involved in influencing cell proliferation and apoptosis in leukemic cell lines. The present studies on chalcones and propanediones herewith, showed that the di-fluorinated propanediones were more potent as compared to their chalcone counterpart. As also, the halogen substituted molecules proved to be more potential.
Biography:
Dr. Ramaa is professor, head of department of pharmaceutical chemistry at Bharati Vidyapeethʼs College of Pharmacy, Navi Mumbai. She received her Ph. D in pharmaceutical chemistry from Institute of chemical technology in 1997 and since then she has been associated with the Bharati Vidyapeethʼs college. She has received several grants from renowned funding agencies like Lady Tata Memorial trust, Basic Research in Nuclear sciences. She has published more than twenty five research and review articles in international and national esteemed journals. She has also presented more than thirty presentations at national and international conferences. She has recently edited a special issue on drug reprofiling in Current topics in medicinal chemistry, Bentham sciences.
Drug Discovery Program at Ontario Institute for Cancer Research, Canada
Department of Pharmacology and Toxicology, University of Toronto, Canada
Histone methylation is a key component of epigenetic signaling and transcriptional regulation. The Mixed Lineage Leukemia (MLL) genes encode a family of histone methyltransferases that activate gene expression through the methylation of histone H3 on lysine 4 (H3K4). Rearrangement and amplification of the MLL1 locus are drivers of leukemogenesis, accounting for 10% of AML in adults and nearly 70% of ALL in infants. In addition, MLL1 mutations are common in a variety of solid cancers, including breast, colon, lung, and bladder. WD40 repeat protein 5 (WDR5) is a component of the multiprotein MLL1 complex that is essential for its methyltransferase activity, and disruption of the WDR5/MLL1 interaction may therefore present a viable therapeutic option for the treatment of MLL-dependent leukemias.
We conducted a medium throughput screen that identified micromolar drug like hits. Following several rounds of optimization using a structure-based approach and focused virtual library design we were able to identify OICR-9429, a nanomolar, highly selective and cell permeable inhibitor of the WDR5-MLL interaction. OICR-9429 is the first highly potent small molecule antagonist of the WDR5-MLL1 interaction and will serve as a valuable molecular probe for further exploration of WDR5 function.
Biography:
Dr. Al-awar earned a PhD in synthetic organic chemistry from North Carolina State University working on Lycopodium Alkaloids and did a postdoctoral fellowship focused on natural products synthesis at the University of North Carolina at Chapel Hill prior to joining Eli Lilly and Company in 1995. While at Lilly, she was an active medicinal chemist in the oncology area working in multidisciplinary teams on the antimicrotubule agent Cryptophycin and later on several kinase focused efforts. In 2002, while at Lilly, Dr. Al-awar took on administrative responsibilities as Head in Discovery Chemistry Research and Technologies and later as Head, Route Selection, in Chemical Product Research and Development prior to joining the Ontario Institute for Cancer Research to build a Drug Discovery Program in July 2008.
School of Pharmaceutical Sciences of Ribeirão Preto, University of São Paulo, Brazil
Nowadays, the biggest challenge for the chemotherapeutic treatments and which is responsible for many cases of failures in both the cancer and the leishmaniasis treatments is called multidrug resistance (MDR). MDR is not only related to a class of medication or to a specific target, but it can also be related to the multiple factors involved in this process. The pathways that affect the decrease of drug concentration in the intracellular environment are related to the decrease associated with the inflow of the carriers, e.g. a diminution via the ABC superfamily (ATP-binding cassette) through the efflux caused by the action of P-glycoprotein (MDR1). Within the ABC superfamily, the P-glycoprotein (P-gp) is the most widely studied class and the calcium channel blockers class is the first generation of P-gp modulators. Moreover, they interfere with the adhesion of the parasite to macrophages and this could be an important strategy to control the initial phase of leishmaniasis.
In this work, we carried out the design of drug candidates addressed to be selective against tumor cells, provided with antiproliferative and leishmanicidal properties through inhibition of tubulin, and not suffering the MDR phenomenon. We have used Ligand-Based Drug Design (LBDD), Structure-Based Drug Design (SBDD), pharmacokinetic and toxicological (ADME/Tox) predictions, as well as homology modeling of a L-type calcium channel. Resulting model was well evaluated as the top- ranked amongst three other models based on two other structures detected in a previous Blastp search. The two drug design approaches above mentioned were then used to perform virtual screening in different commercial compounds databases, totalizing 15-20 compounds selected for purchase. In silico validations of the approaches here used were initially performed using small “contaminated” (with known active ones) databases, and our drug candidates were then validated in vitro, showing interesting bioactivity.
Biography:
Carlos Tomich is Associate Professor of Medicinal Chemistry, at School of Pharmaceutical Sciences of Ribeirão Preto (Brazil). He was born in Brazil, where he did all his studies including Chemistry (State University of Campinas), Masterʼs (Military Engineering Institute), Ph.D. (University of São Paulo), two postdoctoral stages including the Barcelona Biomedical Research Park – Spain, at the Dr. Manuel Pastorʼs group. In research, he works with Computational Medicinal Chemistry, with particular emphasis in cancer, Alzheimerʼs and inflammatory diseases. He has 1 patent and 137 publications (112 papers, 25 book chapters). He is also editor of 4 international books of Medicinal Chemistry.
Chemistry Department, Faculty of Science, South Valley University, Egypt
Carbon paste electrodes have been modified with some lipids for the sensitive and selective detection of the antidepressant (Desipramine). Voltammetric experimental conditions were optimized taking into account the importance of quantifying desipramine in the complex media and in the pharmaceutical formulations. The sensor(Lauric acid modified carbon paste electrode) responds to desipramine giving a cathodic current (at +0.88 V vs. Ag/AgCl electrode and pH 9). The response was characterized with respect to preconcentration potential, accumulation time, paste composition, possible interferences and other variables. A linear relationship between peak response and desipramine concentration over the range from 1 x10-7 to 1 x10-6 M. with standard deviation of 5.5 %. A detection limit of 3.3 x10-10M was obtained under the optimum conditions The method has been applied to the determination of desipramine in serum and urine samples.
Biography:
Mahmoud Khodari Maeila Hamed is a Prof. of Analytical chemistry in South Valley University, Qena, Egypt. His education is B.SC. (general chemistry) at May 1980 Assiut University, Egypt, M.Sc. (Analytical chemistry) 1985, Assiut University and Ph.D at (Analytical Chemistry) 1990, ULB, Belgium- Assiut University, Egypt. His Publications are 54 in the field of analytical chemistry, drug analysis. He attended 21 conferences and meetings.
1Institute of Organic Chemistry and Biochemistry, Czech Academy of Sciences, Czech Republic
2Rega Institute for Medical Research, Katholieke Universiteit Leuven, Belgium
Acyclic nucleoside phosphonates (ANPs) belong to the most successful research topics in our Institute. Systematic investigations in this area resulted in three crucial antiviral drugs in medical practice: cidofovir, adefovir and tenofovir. Entering tenofovir and its therapeutic combinations (Truvada, Atripla, Complera, Stribild) to the market has the lionʼs share on transformation of HIV/AIDS from the life-threatening emergency to a manageble chronic disease.
Ourpresent research in the field is targeted not only to synthesis of new structures but also to improvement of pharmacokinetic properties of compounds already known. There aredozens of therapeutically attractive ANP structures never advanced to the stage of preclinical/clinical investigations. These compounds have usually sufficientantiviral activity but very low bioavailability caused by their polar character. The way to overcome this problem is synthesis of prodrugs. In this work, we present syntheses of various structural types of ANP prodrugs and comparison of their antiviral activities. These prodrugs are: amino acid ester phosphoramidates, POM and POC esters, alkyl and alkoxyalkyl esters, salicylic esters and (methyl-2-oxo-1,3-dioxol-4-yl) methylesters. We focus namely to two pharmacologically interesting ANP types: 2,4-diamino-6-[2- (phosphonomethoxy)ethoxy]pyrimidines– so-called “open-ring” derivatives and to the group of 5-azacytosine acyclic nucleoside phosphonates, especially1-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]-5-azacytosine (HPMP-5-azaC) and1-[2- (phosphonomethoxy)ethyl]-5-azacytosine (PME-azaC). HPMP-5-azaC and its cyclic form have been developed in our group as less toxic and more effective alternatives to cidofovir.
The prodrug form, hexadecyloxyethyl ester of cyclic HPMP-5-azaC revealed the highest values of anti-DNA virus activities including imposing selectivity indices on the order of thousands, e.g. 1160 for herpes simplex virus (HSV-1), ≥5800 for varicella zoster virus (VZV) or ≥24600 for human cytomegalovirus (HCMV). The only disadvantage of HPMP-5-azaC is its complicated metabolic profile due to instability of the 5-azacytosine ring in alkaline conditions including physiological pH. Comparing stability of various 5-azacytosine ANPs, we found much better stability for another 5-azacytosine derivative, 1-[2-(phosphonomethoxy) ethyl]-5-azacytosine (PME-azaC). Despite the fact that antiviral activity of this free phosphonic acid was only marginalwe found promising antiviral activity in its prodrugs, especially against VZV and HCMV. It is another proof that some acyclic nucleoside phosphonates can increase their activities after transformation to prodrugs. Moreover, in some cases not only activity values but also the spectrum of activities could be influenced by transformation to prodrugs.
This work was supported by the Subvention for development of research organization RVO 61388963 and the grant 14-00522S by the Grant Agency of the Czech Republic.
Biography:
Dr. Marcela Krečmerová studied organic chemistry at Charles University in Prague and defended her PhD. thesis at the Institute of Organic Chemistry and Biochemistry (IOCB) of the Czech Academy of Sciences in 1990. Most of her scientific cereer she has been working in the field of nucleoside and nucleotide analogues, many years in a close collaboration with Professor Antonín Holý, the founder of acyclic nucleoside phosphonate chemistry. Her research in the field of 5-azacytosine compounds resulted in development of a new class of extraordinary activeantivirals against DNA viruses. Recent investigations of Dr. Krecmerova are focused to development of prodrugs of biologically active molecules to improve their pharmacological properties and enable their future development as potential drugs. In 2012-2015 she was a head of the junior researchteam at the IOCB focused on developmentof novel therapeutics for cancer and viral diseases. She holds two patents and has co-authored 55 original scientific papers.
1Chemistry Department, College of Science, Sultan Qaboos University, Oman
2Biomolecular NMR, Ruhr University of Bochum, Germany
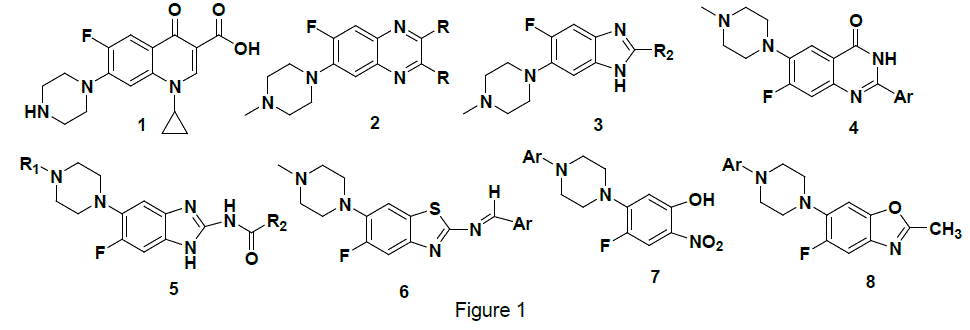
The chemistry of heterocycles has emerged as a fundamental division of organic synthesis which is highly contributed to drug discovery. Structural modification of heterocycles comprises at least half of all organic chemistry research worldwide. Benzazoles, as one class of heterocycles, form the basis of many pharmaceutical, agrochemicals, veterinary, and natural products. Moreover, benzazole derivatives possess a diverse range of biological activities, which make them a good skeletons for drug design. The deisgned introduction of substituents on benzoazole can dramatically alter its biological activity. Fluorine and piperazine are common appendages in valuable therapeutic molecules in medicinal chemistry.
For instance, ciprofloxacin® (1) is a powerful antibiotic with a broad spectrum of biological activity and its activity was related to the incorporation of fluorine and piperazine1. Previously, we have successfully synthesized several heterocycles2-4(2-4) and recently benzoazoles5(5-8) bearing fluorine and piperazine as shown in Figure 1.
Biography:
Abdel-Jalil, R. J. is Associate Professor in organic chemistry at the Department of Chemistry at Sultan Qaboos University. He was a postdoctoral research fellow in Crump Institute for Molecular Imaging at UCLA. Abdel-Jalil, R.J. earned his doctoral degree in Organic Chemistry at Tuebingen University, Germany under the auspices of German Academic Exchange Service (DAAD) Fellowship. He obtained a Master of Science degree and a Bachelor of Chemistry degree from the University of Jordan, Jordan. Abdel-Jalil is the authored/co-authored of over 40-refereed scientific articles as of May 2016. His research interests include development of new methods in organic synthesis, synthesis of biological active compounds and chemical modification of stationary phases. Abdel-Jalil has developed several new methods in chiral and achiral synthesis of heterocyclic systems.
Nizamʼs Institute of Medical Sciences (NIMS), India
Background: Diabetes is associated with an increase risk of atherosclerosis including platelet hyper-reactivity increased inflammation, and endothelial dysfunction. Terminalia chebula (TC) is reported to possess antidiabetic, anti-oxidant, anti-inflammatory activity amongst others. The present study was thus undertaken to evaluate the effect of TC 250mg, TC 500mg versus Placebo on endothelial function in patients with type 2 diabetes.
Methodology: After IEC approval and informed consent, eligible subjects were randomised to either–(1) one capsule of TC 250mg, (2) one capsule TC 500mg or (3)one capsule of Placebo, each given twice daily for 12 weeks. Subjects were reviewed at 4 weeks, 8 and 12 weeks of therapy. At each visit they were evaluated for efficacy and safety. Pharmacodynamic evaluation for endothelial function (change in RI) done at every visit. Blood samples were collected for evaluation of biomarkers (Nitric oxide MDA, Glutathione, hsCRP) Lipids and safety lab tests before and at end of treatment. ANOVA and t Test were used for statistical analysis by Prism Graphpad.
Results: Of 74 screened, total 60 eligible subjects completed the study, twenty in each group. Significant reduction in RI was noted with TC 250mg (−2.38±0.82% to −4.93±1.87% ; p<0.001) and TC 500mg (−2.35%±0.85% to −6.14%±1.01%;p<0.001) suggesting improvement in endothelial function compared to baseline and placebo (-2.11+1.61 to -1.01+2.05%). Significant improvement noted in biomarkers of oxidative stress and systemic inflammation with TC 250 and 500mg compared to baseline and placebo. All treatments were well tolerated.
Conclusion: Both TC250 and 500mg significantly improved endothelial function and reduced biomarkers of oxidative stress without any significant changes in safety labparameters. Terminalia chebula 500mg twice daily produced more pronounced response on pharmacodynamic parameters of endothelial function and biomarkers of oxidative stress as evidenced by significant reduction in mean RI index and improvement in nitric oxide, Glutathione and hsCRP compared to TC 250mg and placebo.
Biography:
DNB (Clinical Pharmacology); MD (Pharmacol); FCCP; FIPS; PG Diploma Bioethics
Usharani Pingali certified by NABH Accreditation board for accreditation of Clinical trials site, Ethics Committees and Investigators. She has more than 20 years of experience in clinical research. Guide to MD, DM and PhD students. She received a number of scientific awards including LK Oration in Sept 2012, she got UK Seth Gold medal for best research paper in clinical pharmacology; PP Suryakumari Medal for best research publication in diabetes, Ford Foundation Travel Fellowship, Minifellowship On Lipid Disorders training at Glastone Institute of Cardiovascular Sciences and Ati Vishisht Chikitsa Gold Medal from Association of College of Chest Physicians, New Delhi. Her fields of interests are Exploratory Phase I studies of new molecules, Developed non-invasive methods to evaluate the pharmacodynamic effects of drugs in early drug development phase, Scientific evaluation of drugs from herbal origin, Pharmacodynamic evaluation of drugs on endothelial function, Evaluation of drugs in diabetes, dyslipidemia and hypertension, Quality Assurance and Audit of clinical research projects.
Specialized Area of Work: Worked in field of clinical pharmacodynamics for over 20 years with specific interest in evaluation of pharmacodynamic effect of drugs using non-invasive methods especially in relation to endothelial dysfunction. Developed and validated some techniques to study drug effects and mechanism of action of drugs especially on Endothelial function and cardiovascular effects, psychomotor performance, gastro-intestinal motility, pain, saccadic eye movement and other CNS effects etc. Worked extensively on herbal formulations also as there are very few systematically conducted studies on the pharmacodynamic effects. Instrumental in establishing ICMR Advanced Centre for Clinical Pharmacodynamics.
1Xuzhou Medical University, China
2 Leiden Academic Centre for Drug Research (LACDR), Leiden University, The Netherlands
In the past decade drug research community has started to appreciate the indispensable role of ligand-receptor binding kinetics (BK) in drug discovery. Next to the classical equilibrium-based drug evaluation process with affinity and potency values as outcomes, kinetic investigation of the ligand-receptor interaction can aid compound triage in the hit-to-lead campaign and provide additional information to understand the molecular mechanism of drug action. To this end, we performed an extensive structure-kinetics relationship (SKR) study in addition to a traditional structure-activity relationships (SAR) analysis at a prototypical drug target—the adenosine A2A receptor (A2AR). The ensemble of 24 antagonistsdisplayed only minor differences in affinity, while they varied substantially in their binding kinetics at the target. Such a combination of SKR and SAR analysis will have general importance for medicinal chemistry efforts on other drug targets as well.
Biography:
Dong Guo, PhD obtained his PhD degree from Leiden University, The Netherlands, in 2014. Since then, he has worked as a postdoc fellow at the same university within the K4DD (Kinetics for Drug Discovery) consortium, financially supported by Europeʼs Innovative Medicines Initiative (IMI) program and major Pharma companies. In February 2016, he joined the Jiangsu Key Laboratory of New Drug Research and Clinical Pharmacy at Xuzhou Medical Universityin China. His main research interest is the investigation of ligand-receptor binding kinetics at G protein-coupled receptors, from both theoretical and experimental perspectives.
Taif University, Kingdom of Saudi Arabia
Consultant of forensic chemistry, Medicolegal institute, Egypt
Expert of Narcotics and Drugs, USA
The present work aims to synthesize novel diclofenac derivatives containing L-alanine moiety. The synthesized compounds docked into the active site to discover validated inhibitors of cyclooxygenases (COX-1 and COX-2). The calculations in-silico were predicted that, the compound with lowest energy of docked poses was interacted with residues of active site, perhaps could be making them possible selective inhibitors against (COX-2) and physiologically active. The binding score of compound compared with reference drug, and show extensive interactions with the targets, which may consider it a suitable selective inhibitor against (COX-2).
Keywords: ALANINE, DICLOFENAC, COX, DOCKING, ADMET.
Biography:
Magda Hassan Abd El lattif is an assistant professor of pharmaceutical organic chemistry in Pharmacy college, Head chairman of pharmaceutical chemistry department, Deanship of scientific research at Taif University, Kingdom of Saudi Arabia. She is also a Consultant of forensic chemistry, Medicolegal institute, Egypt and also is an Expert of Narcotics and Drugs, Tiaft, USA.
University of the Western Cape, South Africa
In this study we described a potential isoniazid (INH) biosensor platform based on zinc oxide doped- poly(8-anilino-1-napthlene sulphonic acid) (ZnO/PANSA) as a diffusional mediator and N-acetyltransferase (NAT2) on gold electrodes. To acknowledge the health complications amongst TB diagnosed patients as a result of the inappropriate dosing of isoniazid; devices with fast response times with enhanced performances and increased sensitivities are essential. This study reports the synthesis and characterization of electroactive platforms for application in the development of nanobiosensors suitable for the appropriate dosing of clinically diagnosed patients by promptly quantifying the levels of the TB drug Isonaizid.
Biography:
Dr Ajayi is a senior lecturer at the Chemistry Department and a research leader at the Enzyme Sensor Laboratory, Sensor Lab at the University of the Western Cape (UWC). She teaches Physical Chemistry at second, third year and honours level and is involved in postgraduate research supervisions. At Sensor Lab she specializes in research that involves the development of drug (particularly HIV and TB treatment drugs) metabolism biosensors and the synthesis of various conducting polymeric and metallic nanomaterials. Dr Ajayi is the recipient of various grant awards and has research collaborations in South Africa, France and the US.
Contact us for any additional information - contact@madridge.org
 Madridge Publishers is licensed under a Creative Commons Attribution 4.0 International License.
Madridge Publishers is licensed under a Creative Commons Attribution 4.0 International License.