

Review Article
Cholestasis in Newborns and Infants
1Fellow of the Paediatric Gastroenterology Program, Faculty of Medicine, University of Chile, Chile; Gastroenterology Department, Hosp. A. Riffart de Castro, Chile
2Institute of Nutrition and Food Technology (INTA), University of Chile, Chile
3Division of Pediatrics, Pontificia Universidad Católica de Chile, Santiago, Chile; Pediatric Gastroenterology Unit, Hospital Sotero del Río, Chile
*Corresponding author: Fernando R. Cáceres, University of Chile, Macul 5540, Santiago, Chile, E-mail: frcv21@yahoo.com
Received: April 07, 2022 Accepted: April 28, 2022 Published: May 5, 2022
Citation: Cáceres FR, Peña A, Araya M. Cholestasis in Newborns and Infants. Int J Pediatr Neonatal Prim Care. 2022; 2(1): 28-33. doi: 10.18689/ijpn-1000108
Copyright: © 2022 The Author(s). This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Cholestatic jaundice early in life is infrequent but always abnormal and must be investigated because early diagnosis is crucial to identify treatable diseases, improving the outcome and quality of life of patients. Some diagnostic tools are efficient but expensive and frequently they are not available everywhere, making difficult to reach universally applicable algorithms. In fact, criteria available in the literature often depend on the facilities of the centres where studies are performed. This explains why until today clinical assessment and frequent clinical check-ups early in life remain being main diagnostic tools for a timely diagnosis and management. This is a narrative review updates the state of the art on neonatal cholestasis and based on the evidence gathered a diagnostic approach is proposed., which considers alternatives depending on the local capacities, emphasizing the need of early recognition of altered stools and conjugated bilirubin, and opportune referral to specialized centres.
Keywords: Cholestasis, Jaundice, Biliary Atresia
Introduction
Cholestasis in infancy is a relevant problem present in 1:250 term newborns that can present serious consequences if not timely treated. Most often, neonatal jaundice is due to indirect/unconjugated bilirubin and resolves spontaneously. However, persistent jaundice is abnormal and may be the earliest sign of severe hepatobiliary or metabolic dysfunction. Among the numerous types of cholestasis, biliary atresia (BA) represents the most frequent cause of liver transplant [1]; its frequency and the number of Kasai procedures required seems to be on the rise. Delayed diagnosis is one of the most relevant factors explaining poor prognosis[1,2]. We here review the most frequent causes, diagnostic criteria, and management of cholestasis with the purpose of identifying the factors that obstruct and delay the diagnostic process and we propose an algorithm that may help improving well-timed diagnosis and treatment of affected patients.
1. Definition and Main Causes of Cholestasis
Definition
Cholestasis refers to the reduced bile production or flow secreted into the intestinal lumen. In neonates is often difficult to differentiate physiological jaundice from the other causes that produce liver injury. Jaundice becomes clinically apparent only when total serum bilirubin exceeds 2.5- 3.0. mg/dL; therefore, early measurement of total and direct serum bilirubin is relevant in jaundiced newborns. The North American Society for Pediatric Gastroenterology, Hepatology & Nutrition (NASPGHAN) and the European Society for Pediatric Gastroenterology and the Hepatology and Nutrition (ESPGHAN, 2018)[2], currently define cholestasis at 1mg/dL direct/conjugated serum bilirubin. The definition was modified because bilirubin excretion into canaliculi may limit the general clearance speed and high non-conjugated bilirubin can retain some of the conjugated bilirubin. Diazo and van den Berg techniques are the usual methods applied[3] to measure direct bilirubin (conjugated plus delta or albumin bound bilirubin).
Causes of Cholestasis
These are diverse and numerous (Table 1); most studies focus on extrahepatic BA, a relevant condition that although infrequent (estimated incidence of 1:5,000 live births), early surgical treatment is efficacious and prevent liver failure and death. Although there are several causes of cholestasis in addition to extrahepatic BA (Table 1), their descriptions are few and relative incidence is low [4].
2. Diagnostic Approach Cholestasis
Diagnostic Approach in Newborns and Infants
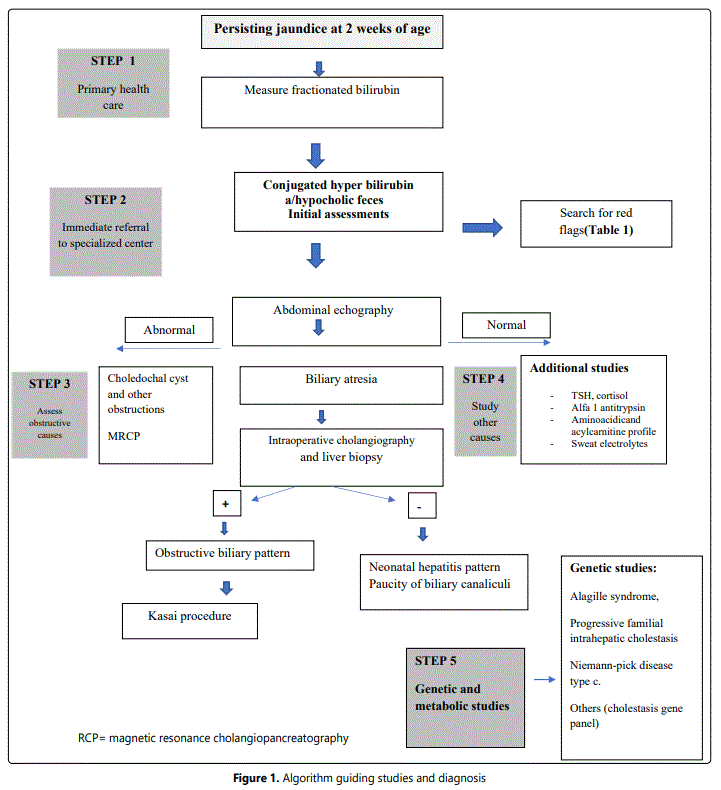
Theoretical consensus seems easy, but in daily life protocols to approach the young, jaundiced patients vary considerably due to variable local difficulties to reach specialists and different local laboratory capacities to support the studies required for diagnosis. Thus, although guidelines and protocols are available, the diagnostic process is often jeopardized by local limitations [5-7]. In the next paragraphs we describe a step-by-step approach to be applied beginning at the primary health system and/or pediatric center (where first postnatal checkups occur), followed by the specialized studies in centers where diagnosis is confirmed and surgical treatment is provided, when possible. Table 2 shows the alert signs, or “red flags” revealed by the clinical history, which help directing initial diagnosis. Figure 1 proposes a diagnostic algorithm that summarizes the experiences obtained from the literature review[8,9].
Step 1: Fractionated bilirubin must be requested to all jaundiced infant at two weeks of life. This must take place during the first routine checkups performed at the primary health care level; therefore, being alert becomes critical [10, 11]. Bilirubin >1 mg/d direct/conjugated serum bilirubinmeans cholestasis and the study protocol must be initiated[2]. Stool assessment is most helpful[12, 13].Evaluation at one month of age using the stool color chart helps confirming acholic stools, which characterizes obstructive biliary conditions like BA[14]. If cholestasis is confirmed, the patient must be referred for assessment by a specialist. Referral must not be delayed since prognosis depends on early treatment. This is the fundamental step in the early diagnostic process. It depends on the clinical evaluations, and all members of the health team (including nurses, midwives, and physicians) must be involved in first suspecting jaundice and feces with altered color that deserve further study[15].
Step 2: This step characterizes by the assessment made by a specialist. The clinical history is crucial to suggest likely etiologies [16], potential anomalies and red flags (table 2). Initial laboratory tests include liver function tests (AST, ALT,INR, albumin, glycemia), which may reveal acute liver failure; and GGT, which when elevated suggests an obstructive condition and when normal or low may be secondary to progressive familial intrahepatic cholestasis (PFIC) or poor biliary acids synthesis[17]. Whole blood count and urinary tests help searching for acute or urinary infections and TSH y T4 assesses hypothyroidism. In this step is important to diagnose/reject neonatal liver insufficiency, severe bacterial infection, herpes infection, neonatal alloimmune hepatitis, galactosemia and mitochondrial diseases; all these conditions must be immediately treated, when possible[18].
Step 3: An abdominal echography must be obtained[19, 20]. Collapsed or absent gallbladder or triangular cord sign are all characteristics suggesting BA and their presence should rapidly lead to an intra operative cholangiography with liver biopsy and subsequent Kasai portoenterostomy, as needed [21-25].
If the abdominal echography shows other alterations, a cholangial resonance helps demonstrating a cyst, a biliary tract tumor, pancreatic malformations, and others. Although a normal echography does not rule out BA. Investigations searching for other diagnoses should start at this point[26].
Step 4: This phase becomes important when BA is overruled; other diagnoses must be sought, such as metabolic conditions [5, 27-33]; acyl carnitine and aminoacidic profile; urinary succinyl acetone and galactose 1PUDPG will help assessing urea cycle disorders, tyrosinemia and galactosemia among others. The infectious diseases screening is relevant when viral, bacterial, and parasitic liver infections are suspected (CMV, herpes simplex virus type 1 and 2, VHB, VHC, VEB, GBS sepsis, toxoplasmosis, rubella, Chagas disease). The immune reactive trypsinogen and sweat electrolytestests help detecting cystic fibrosis and abnormal alpha 1 antitrypsin levelwith Alpha1-antitrypsin (AAT) deficiency. Other studies include assessment of Alagille Syndrome, dysmorphic facial features, ocular assessment (posterior embryotoxon), spine radiographic evaluation (butterfly vertebrae), and cardiovascular assessment (congenital heart defects)[34].
Step 5: If the diagnosis is still unclear, the next step consists of genetic studies[35]. For many years, despite the adequate blood studies and liver biopsies performed, some cases remained undiagnosed. Today, genetic studies have revealed that there are several genetic conditions that may overlap, expressing different clinical phenotypes. Examples of these are JAG1 and NOTCH2 (Alagille syndrome), ATP8B1 (PFIC type 1), ABCB11 (PFIC type 2), ABCB4 (PFIC type 3), SERPINA1 (A1AT deficiency), ABCC2 (Dubin-Johnson syndrome) and SLC25A13 (Neonatal intrahepatic cholestasis caused by citrine deficiency (NICCD), among others[36]. The most relevant limitation at this stage is that genetic studies are expensive and not available everywhere[37, 38] (Figure 1).
Management of Cholestasis
This firstly refers to differentiate conditions that require early surgical procedures (BA, choledochal cyst) from those that have non-surgical specific treatments (infections, galactosemia, tyrosinemia, hypothyroidism, neonatal alloimmune hepatitis). In those where the primary cause cannot be modified, medical management will mainly aim at treating chronic complications (fat malabsorption, vitamin deficiencies, pruritus, hypercholesterolemia, cirrhosis, portal hypertension, liver insufficiency) and avoid nutritional deterioration. Adequate medical management maintains the best possible quality of life of patients and must start as soon as diagnosis is reached[39]. The administration of albumin and immunoglobulin can be useful in cases of neonatal alloimmune hepatitis and the use of glucocorticoids in biliary tract atresia. Nutritional support is usually necessary to minimize failure to thrive; fat-soluble vitamins, phenobarbital, ursodeoxycholic, rifampicin, cholestyramine may be helpful tools for management, but one must keep in mind that specific treatment of the different cholestatic conditions must be supervised by a specialist[40].
Discussion
The evidence reviewed confirms the diversity of protocols applied to study, diagnose and manage cholestasis in neonates and infants[41]. The algorithm proposed (Figure 1) appears as a useful tool that considers the variable capacities available in the different health systems levels in many countries. Although guidelines and recommendations are available, delayed treatment continues being the main cause of poor prognosis in cases in which surgical management is possible[42]. Difficulties in recognizing truly acholic faeces early in life, access to trained specialists and the possibility to perform expensive tests are the main limitations identified. Modification of historic cholestasis definition by NASPGHAN and ESPGHAN [2] is relevant and improves the chance of early diagnosis. Measuring fractionated bilirubin at two weeks of age must be stressed. In fully breast-fed babies, who lack risk factors or red flags this measurement could be delayed until the third week of life but, certainly the criteria to keep in mind is that fractionated bilirubin should be measured at two weeks of age.
Probably the most frequent and difficult task in daily practice is to identify significant changes in stool pigmentation (normal, hypocholic or acholic). Parents and health professionals tend to evaluate stool pigmentation subjectively, with frequent erroneous interpretations; for instance, Bakshiet al. determined that almost 40% of doctors and nurses did not recognize altered stools as abnormal[12]. A stool pigment chart is available to help evaluation and using this chart in Taiwan, Shan-Ming Chen et al. obtained 95.2% sensitivity[4]. Yan-Hong Gu et al. conducted a 19-year study using the same chart and found that BA was diagnosed earlier and long-term survival of the native liver was greater[13]; however, this kind of charts in most countries are ignored and not utilized.
Studies assessing the criteria applied for decision making differ in their results, probably influenced by the conditions of the different studies performed, but also, recommendations available somewhat disagree with the evidence reported. The epidemiology and diagnostic results reported in two cohorts, assessed before and after year 2000 described that age of detection diminished from a median of 43 days to 22 days and this resulted in better survival rates[5]. These results are significant and show that it is possible to do better, but unfortunately early diagnosis at 22 days of life is not representative of the typical mean time to reach diagnosis everywhere. Assessment of attitudes and behaviour of 116 participants that provided medical care in primary paediatric systems[41] showed that 94.8% felt confident to correctly diagnose hyperbilirubinemia, though only 10.3% knew the biochemistry of direct bilirubin; of 56% participants that stated to know the “current criteria in use”, only 18.5% answered that the criteria to define cholestasis recently changed. Studies often emphasize the use of certain tests that are expensive and not always available[31]; in contrast, in most studies reviewed the relevance of clinical history and the benefits of using standardized protocols are ignored[43]. In a recent survey applied to paediatricians, most of them answered that they would order measuring fractionated bilirubin at 4 weeks of life, but many did not program the visits needed to perform such measurement and 25% did not see the baby between week 1 and 8 of life[44].
The relevance of early diagnose when surgery is possible is shown by a French study that assessed 695 BA patients and found that the native liver had better survival when the Kasai procedure took place during the first 30 days of life[22]. To improve the situation, predictive models for initial assessment were developed based on hierarchic classification and regression tree (CART) or by logistic regression[21]. Both methodologies failed; 12% of BA babies were erroneously classified as non-BA and 17% of non-BA patients classified as BA. Thus, clinicalhistory and physical examination remain being relevant tools when first assessing the patients and his/her stool characteristics[45]. Fractionated bilirubin, blood screening and imaging assessment are crucial in the next steps.


Conclusion
Cholestatic jaundice that lasts more than the first two weeks of life must be studied because it may be the first sign of severe liver disease. Early detection and confirmation of jaundice and altered colored fecesat the primary health care level and subsequent diagnostic assessment by a specialist are essential for early diagnosis and treatment that result in better prognosis. Specialists are necessary to guide the studies, imaging, liver biopsy and the genetic studies (Figure 1); these guide subsequent studies searching for treatable underlying causes like infections (sepsis), various metabolic and genetic diseases, etc. All studies reviewed agree on that ultrasound is one of the useful initial tests to be performed. Independent of causes, a precise and predetermined approach is the fundamental piece in the process of diagnosis and treatment, and nutritional support is relevant to maintain the best condition possible in the patient.Up to date, variability on how to conduct diagnostic studies adjusted to local limitations remains being the main challenge to obtain better outcomes. The algorithm proposed (Figure 1) considers the potential restrictions locally present, describing the steps to follow at the different health service levels. We emphasize the importance of the primary health level in early detection and timely referral.
Acknowledgements/funding: No external funding.
Conflict of interests: Authors have no conflict of interest to declare.
Ethics Approval: Not applicable
COI: Not applicable
Authorship: All three authors contributed to the conception of the work; interpretation of data; critically analysis and drafting. FC was responsible for the literature review. FC, AP, and MA wrote the drafts and MA is responsible for final manuscript. All authors approved the final version to be published.
References
- De Bruyne R, et al. Clinical practice: neonatal cholestasis. Eur J Pediatr. 2011; 170(3): 279-84. doi: 10.1007/s00431-010-1363-8
- Fawaz R, Baumann U, Ekong U, Fischler B, et al. Guideline for the Evaluation of Cholestatic Jaundice in Infants: Joint Recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J Pediatr Gastroenterol Nutr. 2017; 64(1): 154-168. doi: 10.1097/MPG.0000000000001334
- Lo SF, Kytzia HJ, Schumann G, Swartzentruber M, Vader HL, et al. Interlaboratory comparison of the Doumas bilirubin reference method. Clin Biochem. 2009; 42(12): 1328-30. doi: 10.1016/j.clinbiochem.2009.05.007
- Chen HL, Wu SH, Hsu SH, Liou BY, et al. Jaundice revisited: recent advances in the diagnosis and treatment of inherited cholestatic liver diseases. J Biomed Sci. 2018; 25(1): 75. doi: 10.1186/s12929-018-0475-8
- Santos Silva E, Almeida A, Frutuoso S, Martins E, et al., Neonatal Cholestasis Over Time: Changes in Epidemiology and Outcome in a Cohort of 154 Patients From a Portuguese Tertiary Center. Front Pediatr. 2020; 8: 351. doi: 10.3389/fped.2020.00351
- Götze T, Blessing H, Grillhösl C, Gerner P, Hoerning A. Neonatal Cholestasis - Differential Diagnoses, Current Diagnostic Procedures, and Treatment. Front Pediatr. 2015; 3: 43. doi: 10.3389/fped.2015.00043
- Feldman AG, RJ Sokol. Neonatal Cholestasis: Updates on Diagnostics, Therapeutics, and Prevention. Neoreviews. 2021; 22(12): e819-e836. doi: 10.1542/neo.22-12-e819
- Cho SJ, Kim GE. A practical approach to the pathology of neonatal cholestatic liver disease. Semin Diagn Pathol. 2019; 36(6): 375-388. doi: 10.1053/j.semdp.2019.07.004
- Santos Silva E, Silva HM, Catarino C, Dias CC, Silva AS, Lopes AI. Neonatal cholestasis: development of a diagnostic decision algorithm from multivariate predictive models. Eur J Pediatr. 2021; 180(5): 1477-1486. doi: 10.1007/s00431-020-03886-z
- Dani C, et al. Italian guidelines for the management and treatment of neonatal cholestasis. Ital J Pediatr. 2015; 41: 69. doi: 10.1186/s13052-015-0178-7
- Brits H, Adendorff J, Huisamen D, Beukes D, Botha K, Herbst H, et al. The prevalence of neonatal jaundice and risk factors in healthy term neonates at National District Hospital in Bloemfontein. Afr J Prim Health Care Fam Med. 2018; 10(1): e1-e6. doi: 10.4102/phcfm.v10i1.1582
- Bakshi B, Sutcliffe A, Akindolie M, Vadamalayan B, John S, Arkley C, et al. How reliably can paediatric professionals identify pale stool from cholestatic newborns? Arch Dis Child Fetal Neonatal Ed. 2012; 97(5): F385-7. doi: 10.1136/fetalneonatal-2010-209700
- Gu YH, Yokoyama K, Mizuta K, Tsuchioka T, Kudo T, Sasaki H, et al. Stool color card screening for early detection of biliary atresia and long-term native liver survival: a 19-year cohort study in Japan. J Pediatr. 2015; 166(4): 897-902.e1. doi: 10.1016/j.jpeds.2014.12.063
- El-Shabrawi MH, Baroudy SR, Hassanin FS, Farag AE, et al. A pilot study of the value of a stool color card as a diagnostic tool for extrahepatic biliary atresia at a single tertiary referral center in a low/middle income country. Arab J Gastroenterol. 2021; 22(1): 61-65. doi: 10.1016/j.ajg.2020.12.004
- Rabbani T, et al. Newborn Screening for Biliary Atresia: a Review of Current Methods. Curr Gastroenterol Rep. 2021; 23(12): 28. doi: 10.1007/s11894-021-00825-2
- Lane E, Murray KF. Neonatal Cholestasis. Pediatr Clin North Am. 2017; 64(3): 21-639. doi: 10.1016/j.pcl.2017.01.006
- Lu FT, Wu JF, Hsu FY, Ni YH, Chang MH, et al. γ-Glutamyl transpeptidase level as a screening marker among diverse etiologies of infantile intrahepatic cholestasis. J Pediatr Gastroenterol Nutr. 2014; 59(6): 695-701. doi: 10.1097/MPG.0000000000000538
- Karpen SJ. Pediatric Cholestasis: Epidemiology, Genetics, Diagnosis, and Current Management. Clin Liver Dis (Hoboken). 2020; 15(3): 115-119.
- Humphrey TM. The role of ultrasound in the investigation of neonatal jaundice. Clin Radio. 2020; 75(11): 815-821. doi: 10.1016/j.crad.2020.01.018
- Mittal V, Saxena AK, Sodhi KS, Thapa BR, Rao KLN, Das A, et al. Role of abdominal sonography in the preoperative diagnosis of extrahepatic biliary atresia in infants younger than 90 days. AJR Am J Roentgenol. 2011; 196(4): W438-45. doi: 10.2214/AJR.10.5180
- Shneider BL, Moore J, Kerkar N, Magee JC, Ye W, Karpen SJ, et al. Initial assessment of the infant with neonatal cholestasis-Is this biliary atresia? PLoS One. 2017; 12(5): e0176275. doi: 10.1371/journal.pone.0176275
- Lakshminarayanan B, Davenport M. Biliary atresia: A comprehensive review. J Autoimmun. 2016; 73: 1-9. doi: 10.1016/j.jaut.2016.06.005
- Agin M, et al. Clues to the diagnosis of biliary atresia in neonatal cholestasis. Turk J Gastroenterol. 2016; 27(1): 37-41. doi: 10.5152/tjg.2015.150379
- Sundaram SS, Mack CL, Feldman AG, Sokol RJ. Biliary atresia: Indications and timing of liver transplantation and optimization of pretransplant care. Liver Transpl. 2017; 23(1): 96-109. doi: 10.1002/lt.24640
- Vij M, Rela M. Biliary atresia: pathology, etiology and pathogenesis. Future Sci OA. 2020; 6(5): FSO466. doi: 10.2144/fsoa-2019-0153
- Yerina SE, Ekong UD. Biliary Atresia/Neonatal Cholestasis: What is in the Horizon? Pediatr Clin North Am. 2021; 68(6): 1333-1341. doi: 10.1016/j.pcl.2021.08.002
- Delemos AS, Friedman LS. Systemic causes of cholestasis. Clin Liver Dis. 2013; 17(2): 301-317. doi: 10.1016/j.cld.2012.11.001
- Hoerning A, Raub S, Dechêne A, Brosch MN, Kathemann S, Hoyer PH, et al. Diversity of disorders causing neonatal cholestasis - the experience of a tertiary pediatric center in Germany. Front Pediatr. 2014; 2: 65. doi: 10.3389/fped.2014.00065
- Ipek M, Aydin M, Zenciroglu A, Gokçe S, Okumus N, Gulaldi NCM. Conjugated hyperbilirubinemia in the neonatal intensive care unit. Turk J Gastroenterol. 2013; 24(5): 406-414. doi: 10.4318/tjg.2013.0553
- Junge N, Goldschmidt I, Wiegandt J, et al. Dubin-Johnson Syndrome as Differential Diagnosis for Neonatal Cholestasis. J Pediatr Gastroenterol Nutr. 2021; 72(5): e105-e111. doi: 10.1097/MPG.0000000000003061
- Khalaf R, Phen C, Karjoo S, Wilsey M. Cholestasis beyond the Neonatal and Infancy Periods. Pediatr Gastroenterol Hepatol Nutr. 2016; 19(1): 1-11. doi: 10.5223/pghn.2016.19.1.1
- Pandita A, Gupta V, Gupta G. Neonatal Cholestasis: A Pandoraʼs Box. Clin Med Insights Pediatr. 2018, 12: 1179556518805412. doi: 10.1177/1179556518805412
- Weiss AK, Vora PV. Conjugated Hyperbilirubinemia in the Neonate and Young Infant. Pediatr Emerg Care. 2018; 34(4): 280-283. doi: 10.1097/PEC.0000000000001467
- Dong C, Zhu HY, Chen YC, Luo XP, Huang ZH. Clinical Assessment of Differential Diagnostic Methods in Infants with Cholestasis due to Biliary Atresia or Non-Biliary Atresia. Curr Med Sci. 2018; 38(1): 137-143. doi: 10.1007/s11596-018-1857-6
- Feldman AG, Sokol RJ. Neonatal cholestasis: emerging molecular diagnostics and potential novel therapeutics. Nat Rev Gastroenterol Hepatol. 2019; 16(6): 346-360. doi: 10.1038/s41575-019-0132-z
- Jeyaraj R, Bounford KM, Ruth N, Lloyd C, MacDonald F, et al. The Genetics of Inherited Cholestatic Disorders in Neonates and Infants: Evolving Challenges. Genes (Basel). 2021; 12(11). doi: 10.3390/genes12111837
- Karpen SJ, Kamath BM, Alexander JJ, Ichetovkin I, Rosenthal P, et al. Use of a Comprehensive 66-Gene Cholestasis Sequencing Panel in 2171 Cholestatic Infants, Children, and Young Adults. J Pediatr Gastroenterol Nutr. 2021; 72(5): 654-660. doi: 10.1097/MPG.0000000000003094
- Hertel PM, Hawthorne K, Kim S, Finegold MJ, Shneider BL, Squires JL, et al. Presentation and Outcomes of Infants with Idiopathic Cholestasis: A Multicenter Prospective Study. J Pediatr Gastroenterol Nutr. 2021; 73(4): 478-484. doi: 10.1097/MPG.0000000000003248
- Catzola A, Vajro P. Management options for cholestatic liver disease in children. Expert Rev Gastroenterol Hepatol. 2017; 11(11): 1019-1030. doi: 10.1080/17474124.2017.1359538
- Kriegermeier A, Green R. Pediatric Cholestatic Liver Disease: Review of Bile Acid Metabolism and Discussion of Current and Emerging Therapies. Front Med (Lausanne). 2020; 7: 149. doi: 10.3389/fmed.2020.00149
- Menz TJ, Herzlinger M, Ross A, Zonfrillo MR. Knowledge, Attitudes, and Behaviors of Pediatric Primary Care Providers on Management of Cholestasis. Glob Pediatr Health. 2019; 6: 2333794X19829757. doi: 10.1177/2333794X19829757
- Srivastava A. Optimizing Care and Outcome of Neonatal Cholestasis: Are We on the Right Track? Indian J Pediatr. 2017; 84(8): 578-579. doi: 10.1007/s12098-017-2389-y
- Mandato C, Zollo G, Vajro P. Cholestatic jaundice in infancy: struggling with many old and new phenotypes. Ital J Pediatr. 2019; 45(1): 83. doi: 10.1186/s13052-019-0679-x
- Palermo JJ, Joerger S, Turmelle Y, Putnam P, Garbutt J. Neonatal cholestasis: opportunities to increase early detection. Acad Pediatr. 2012; 12(4): 283-287. doi: 10.1016/j.acap.2012.03.021
- Hasosah M. Knowledge and Practice of Pediatric Providers Regarding Neonatal Cholestasis in the Western Region of Saudi Arabia. Saudi J Med Med Sci. 2021; 9(3): 248-253. doi: 10.4103/sjmms.sjmms_462_20
