

Research Article
Regioselective C-H Borylation of C (sp2)-H Bond
Institute for Molecular Design and Synthesis, School of Pharmaceutical Science and Technology, Health Science Platform, Tianjin University, Nankai District, Tianjin, China
*Corresponding author: Hong-Liang Li, Institute for Molecular Design and Synthesis, School of Pharmaceutical Science and Technology, Health Science Platform, Tianjin University, Nankai District, Tianjin, China, E-mail: lhl522508@126.com
Received: January 30, 2019 Accepted: February 19, 2019 Published: February 26, 2019
Citation: Jing HQ, Antilla JC, Li HL. Regioselective C-H Borylation of C (sp2)-H Bond. Madridge J Nov Drug Res. 2019; 3(1): 114-119. doi: 10.18689/mjndr-1000117
Copyright: © 2019 The Author(s). This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
C-H activation reactions have become a powerful method to direct functionalization of alkyl, alkenyl, and aryl C-H bonds over the past few decades. Among of them, Iridium catalyzed transformation of aryl C-H bonds to C-B bonds is one of the most useful method. However, a central challenge in these reactions is controlling their site selectivity. Over the past decade, some methods have been developed to accomplish regio-selective C-H borylation by catalysts or substrates modification. In this paper, some methods developed in recent years to realize ortho-, meta-, and para-selective C-H borylation will be summarized and their strategy and mechanism of these methods will be discussed.
Keywords: Iridium; Regioselective; C-H borylation; Non-covalent bond interaction.
Introduction
Carbon-Carbon bond are the molecular “bricks and mortar” from which diverse architectures in living organisms and manmade materials are constructed. In the field of organic chemistry, there are numerous methods for carbon-carbon bond construction, which have been developed, ranging from traditional nucleophilic reaction to metal catalyzed reaction for formation of C-C bond. Among of them, Suzuki-Miyaura reaction has become one of the most powerful method for the construction of C-C bond since its discovery in 1979 [1]. Arylboron is an important reagent for participating in this reaction. In addition, organoboron reagents as versatile intermediates have been extensively used in synthetic chemistry because it can be converted to more complex molecules by further transformations, such as Chan-Lam-Evans coupling, [2-4] and oxidation [5-7]. Therefore, developing a highly effective method for synthesis of organoboron reagents is very desirable.
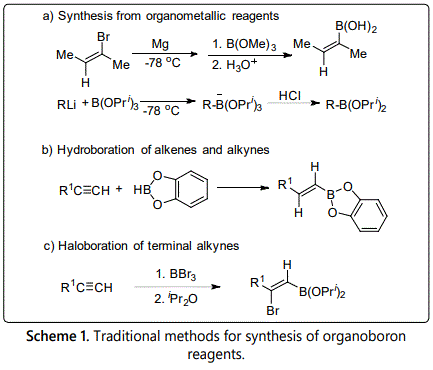
Traditional methods to obtain organoboron reagents are these three methods (Scheme 1), which contains synthesis from organolithium or organomagnesium, hydroboration of alkenes or alkynes and haloboration of terminal alkynes [8]. However, there are some limitations of these methods. For example, the synthesis from organometallics usually requires stoichiometric amounts of strong base such as n-BuLi and a harsh lower temperature to use in the reaction. As for the reactions of hydroboration and haloboration, boron atom always adds to the terminal position of alkenes or alkynes. It is difficult to obtain internal boron reagents form these two methods. Besides, these three methods also are not in accordance with the rule of atomic economy. The development of transition metal catalyzed C-H borylation provides a good way for preparation of organoboron reagents, especially for aromatic boron reagents.
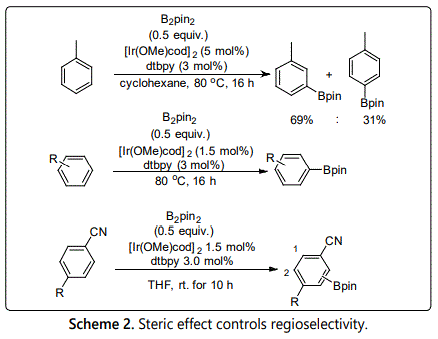
The most active catalyst for the borylation of aromatic compounds is the iridium/bipyridine catalytic system, which was found by Prof. Hartwig, Ishiyama, Miyaura et al. in 2002 [9-11]. This iridium catalytic system could make the C-H borylation of aromatic compounds proceed easily under mild condition. Moreover, they also found the C-H borylation reaction often occurs with regioselectivity controlled predominantly by steric effect [11] (Scheme 2). For instance, mono-substituted arenes as substrates, borylation always gives a mixture of meta- and para-borylated products in 2:1 ratio [9]. The ortho-borylated isomer was usually not formed because the steric hindrance of methyl substituent. In the case of di-substituted arenes, borylation reaction always proceeds at the position with less steric hindrance [12,13]. Reactions of 1, 4-disubstituted aromatic compounds could account for this steric effect more apparently. Borylation of asymmetrically 1, 4-disubstituted substrates, borylation proceeds at the ortho-position of substituent with less steric hindrance between the two possible reactive positions. Although steric effect could control regioselectivity of this reaction, achieving accurately control site-selectivity still exist many challenges in this kind of reaction. In the following, some methods developed in recent years will be summarized according to ortho-, meta- and para-selectivity.
Ortho-Selective C-H Borylation
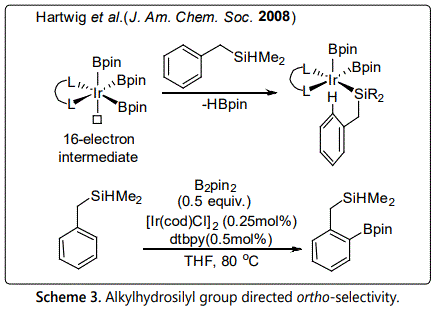
Directing group assist ortho-selective C-H borylation was firstly developed by Boebel et al. in 2008 [14]. They disclosed the example of ortho-selective C-H borylation for aromatic substrates directed by alkylhydrosilyl group (Scheme 3). This reaction proceeded according to a relay directed process. The alkylhydrosilyl group could reversibly attach to iridium center by σ-bond metathesis process to form a 16-electron intermediate, and bring iridium-boryl catalytic species close to ortho-position, which will facilitate the cleavage of ortho C-H bond. Benzyl dimethylsilane as substrate, the reaction could proceed with high ortho-selectivity in good yield.
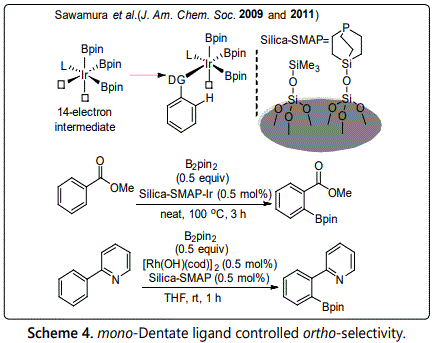
In 2009, Kawamorita et al. [15,16] reported an ortho-selective C-H borylation of aromatic compounds catalyzed by silica supported mono-dentate phosphine ligand. In this reaction, mono-dentate ligand and iridium formed a 14-electrons intermediate, which contains two vacant sites, one for directing group of substrate and the other for the cleavage of ortho C-H bond (Scheme 4). Therefore, the ortho-selective C-H borylation proceeded through the intermediate. The reaction has very wide substrates scope including benzoate, ether, sulfonate and so on. In addition, he also developed an ortho-selective C-H borylation of 2-phenylpyridine in 2011, which was catalyzed by Rh with silica supported mono-dentate phosphine ligand.
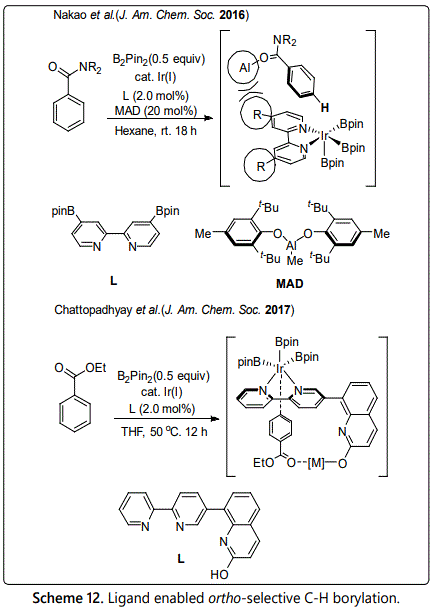
Later, Ligand-enabled ortho-selective C-H borylation was found by several groups, in which ortho-selectivity is achieved by modifying ligand structure (Scheme 5). In traditional methods, bipyridine type ligand is the most commonly used in iridium catalyzed sp2 C-H borylation reaction. In 2014, Ghaffari et al. [17] reported silyl-phosphorous ligand catalyzed ortho-selective C-H borylation of alkyl benzoate. The reaction not only gave good yields of borylated product but also controlled ortho-selectivity very well. In 2016, Bisht and Chattopadhyay [18] also developed ortho-selective C-H borylation of benzaldehydes using 8-aminoquinoline as ligand, in which tert-butylamine was as the traceless protecting/directing group [17,18]. These two ortho-borylations could be suitable for a broad range of substrates and all of them give good to excellent yield and regioselectivity. The mechanism of these two reactions is similar to directing group assisting process. Designed ligands occupy two vacant orbitalʼs, directing group brings the other one close to ortho-position.

In addition, Wang et al. [19] reported a new ortho-selective C-H borylation catalyzed by a designed N, B-bidentate boryl ligand in 2017. Introducing convenient silylborane precursors onto N, B-bidentate boryl ligands, the iridium (III) complex was formed via Si-B oxidative addition.
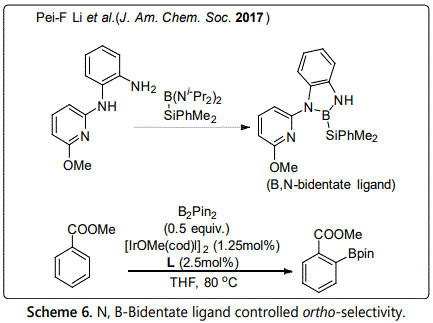
However, no matter in directing group assist ortho-borylation or in ligand-enabled ortho-borylation, all of them usually require already presence or installation of a directing group in substrates. In contrast, traceless directing group would be more attractive alternatives; non-covalent bond interaction controlled regioselective C-H borylation reaction is a new strategy. At present, non-covalent organocatalysis has been successfully applied into some reactions to achieve regioselective C-H borylation of aromatic substrates by employing hydrogen bonding, ion pairing, Lewis acid-base interaction and electrostatic interaction. The first ortho-selective C-H borylation of aromatic compounds controlled by non-covalent bond interaction was found by Smith et al. in 2012, in which hydrogen bonding interaction between the H atom of Boc protected aniline substrate and the O atom of Bpin ligand favored ortho-selective C-H borylation [20].
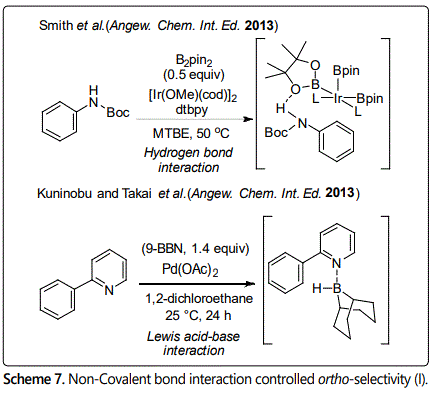
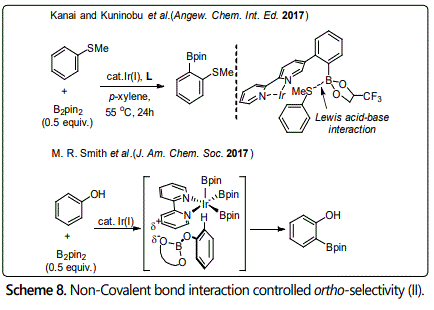
In 2013, Kuninobu [21] reported an ortho-selective C-H borylation of 2-phenylpyridine. In this reaction, 9-borabicyclo[3.3.1] nonane (9-BBN) was selected as boryl reagent; the boryl group was introduced at the ortho-position of 2-phenyl pyridine by Lewis acid-base interaction between the Lewis basic N atom and the Lewis acidic B atom [20]. Based on this result, Li et al. [22] developed an ortho-selective borylation of aryl sulfides using a Lewis acid-base interaction between designed ligand in the catalyst and a substituent (a sulfur atom) of the substrates in 2017 (Scheme 8). They think the steric repulsion between the iridium-boryl catalytic species and substituent(s) of the substrates would be an obstacle to realize ortho-selectivity; it is difficult to promote the site-selectivity using other non-covalent bond interaction such as hydrogen bonding interaction. Thus, a stronger interaction was used in this reaction. Almost the same time; Smith et al. reported a strategy for ortho-selective borylation of phenol derivatives. From selectivity of observation with ArylOBpin (pin = pinacolate) [23], they hypothesized that an electrostatic interaction between the partial negatively charged OBpin group and the partial positively charged bipyridine ligand of the catalyst favors ortho-selectivity.
meta-Selective C-H Borylation
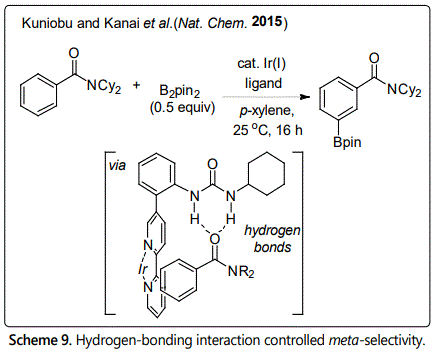
The small difference in intrinsic reactivity of C-H bonds in organo-molecules makes it difficult to achieve regiocontrol. Directing group assist ortho-selectivity by forming five or six cyclometallation has been developed very well in the past years. However, the directed activation of C-H bond that are distal to directing group still exist challenges such as meta- and para-selectivity because the target C-H bond is geometrically inaccessible to directed metalation owing to the ring strain. In this context, non-covalent bond interaction between catalyst species and substrate provide a good way to solve this problem. Recent years, meta- and para-selective C-H borylation of aromatic compounds have been achieved using non-covalent bond interaction. A significant breakthrough was made by Kuninobu group in 2015 [24]. They developed an iridium-catalyzed meta-selective C–H borylation of aromatic compounds using a newly designed catalytic system (Scheme 9). The hydrogen bonding interaction between the urea moiety of designed ligand and a hydrogen-bond acceptor in the substrate (carbonyl of amide) places the iridium catalyst to the meta-position of aromatic amides, eaters, phosphonates, and phosphonic diamide and phosphine oxides. When compared with directing group-controlled reaction, the hydrogen-bonding ligand only required in a catalytic amount which interacts reversibly with the substrate.
In 2016, Davis et al. reported an ion pair-directed approach to controlling regioselectivity in the iridium-catalyzed borylation of two classes of aromatic quaternary ammonium salts [25]. A single electrostatic interaction could be successfully employed to position a reactive metal catalytic species to meta-target C-H bond. This is the first example to demonstrate the viability of ion-paring as a powerful tool for region control.
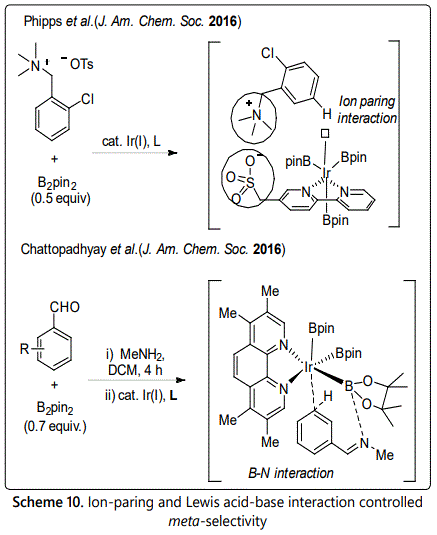
Besides, Bisht reported meta borylation proceeds via an electrostatic interaction and a secondary interaction between a designed ligand and substrate [18]. The origin of meta-selectivity was controlled by two factors: i) an electrostatic interaction of the tris(boryl)iridium complex attached with the electron-rich ligand substrate; ii) a secondary Lewis acid-base interaction between the imine N atom and the boryl B atom of catalyst.
para-Selective C-H Borylation
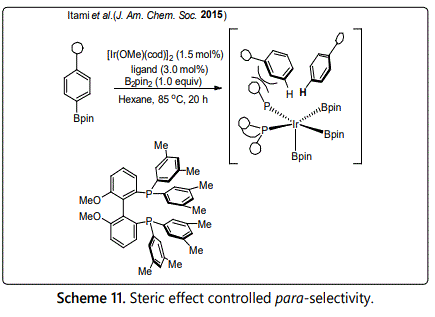
Electronic aromatic substitution is a traditional method to achieve para-selective C-H functionalization. However, the substrate scope of this method is very limit, only when strongly electron-donating groups (EDG) such as a dimethylamino group are attached to the benzene rings. In addition, it always gives a mixture of ortho- and para-product at the same time. Therefore, many groups were devoted into developing a strategy that only favors para-selective functionalization. In 2015, Saito accomplished a highly para-selective aromatic C-H borylation of mono-substituted benzene derivatives by using a new iridium catalyst bearing a bulky diphosphine ligand. The site-selectivity raised from the steric repulsion between the bulkiness of substituent on benzene ring and the diphosphine-iridium catalyst[26].
By using the strategy of steric repulsion between catalyst and substrate, Yang and coworkers reported a method of para-selective C-H borylation of benzamides in 2017 by using cooperative iridium/aluminum catalysis [27]. They thought that the regioselectivity is controlled by the steric repulsion between the coordination of substrate to the bulky aluminum and the boryl-iridium catalytic species because the coordination shields the ortho- and meta- reactive position of substrate (Scheme 11). In addition, Hoque disclosed to a highly efficient method for para-selective borylation [28]. By modifying the core structure of bipyridine, the designed L-shape ligand will recognize the functionality of the oxygen atom of the ester carbonyl group via non-covalent interaction, which provides an unprecedented controlling factor for para-selective C-H activation.
Conclusion
Organo-boron compounds as versatile intermediate has been widely employed in organic synthesis. The development of regioselective C-H borylation provides an efficient way to prepare them using different strategies such as directing group assist ortho-selectivity, ligand enable meta- and para-selectivity. However, there are many challenges that remain to be addressed to improve the practicality and versatility of C-H borylation reaction in the future. For instance, it will be of great importance to develop more effective ligand that enable regioselective C (sp3)-H borylation, especially for remote position. C (sp3)-H bond is building block for constructing natural products, regioselective functionalization could highly improve the efficiency for preparing biological molecules.
Acknowledgment
This work was partially supported by the 64th China Postdoctoral Science Foundation Grant Grant Number 2018M641642.
References
- Maluenda I, Navarro O. Recent development in the Suzuki-Miyaura reaction 2010-2014. Molecules. 2015; 20(5): 7528-7557. doi: 10.3390/molecules20057528
- Chan DM, Monaco KL, Wang RP, Winteres MP. New N- and O-arylations with phenylboronic acids and cupric acetate. Tetrahedron Lett. 1998; 39(19): 2933-2936. doi: 10.1016/S0040-4039(98)00503-6
- Evans DA, Katz JL, West TR. Synthesis of diaryl ethers through the copperpromoted arylation of phenols with arylboronic acids. An expedient synthesis of thyroxine. Tetrahedron Lett. 1998; 39(19): 2937-2940. doi: 10.1016/S0040-4039(98)00502-4
- Lam YS, Clark CG, Saubern S, et al. New aryl/heteroaryl C-N bond crosscoupling reactions via arylboronic acid/cupric acetate arylation. Tetrahedron Lett. 1998; 39: 2941-2944. doi: 10.1016/S0040-4039(98)00504-8
- Simon J, Salzbrunn S, Prakash GK, Petasis NA, Olah GA. Regioselective coversion of arylboronic acid to phenols and subsequent coupling to symmetrical diaryl ethers. J Org Chem. 2001; 66(2): 633-634. doi: 10.1021/jo0015873
- Zhu C, Wang R, Falck JR. Mild and rapid hydroxylation of Aryl/heteroaryl boronic acids and boronate esters with N-oxide. Org Lett. 2012, 14(13): 3494-3497. doi: 10.1021/ol301463c
- Kaewmati P, Somsook E, Dhital NR, Sakurai H. Aerobic oxygenation of phenylboronic acid promoted by thiol derivatives under gold-free condition: a warning against gold nanoparticle catalysis. Tetrahedron Lett. 2012; 53(45): 6104-6106. doi: 10.1016/j.tetlet.2012.08.142
- Miyaura N, Suzuki A. Palladium-catalyzed cross-coupling reaction of organoborn compounds. Chem Rev. 1995; 95(7): 2457-2483. doi: 10.1021/cr00039a007
- Ishiyama T, Takagi, J, Ishida K, Miyaura N, Anastasi NR, Hartwig JF. Mild iridium-catalyzed borylation of arenes. High turnover numbers, room temperature reactions and isolation of a potential intermediate. J Am Chem Soc. 2002; 124(3): 390-391. doi: 10.1021/ja0173019
- Hartwig JF. Regioselectivity of the borylation of alkanes and arenes. Chem Soc Rev. 2011; 40(4).
- Ros A, Fernandez R, Lassaletta JM. Functional group directed C-H borylation. Chem Soc Rev. 2014; 43(10): 3229-3243.
- Ishiyama T, Takagi J, Hartwig JF, Miyaura NA. A stoichiometric aromatic C-H borylation catalyzed by iridium(I)/2,2′-bipyridine complexes at room temperature. Angew Chem Int Ed Engl. 2002; 41(21). doi: 10.1002/1521-3773(20020816)41:16
- Ishiyama T, Nobuta Y, Hartwig JF, Miyaura N. Room temperature borylation of arenes and heteroarenes using stoichiometric amounts of pinacolborane catalyzed by iridium complexes in an inert solvent. Chem Commun. 2004; 35(23): 2924-2925.
- Boebel TA, Hartwig JF. Silyl-directed, iridium-catalyzed ortho-borylation of arenes. A one-pot ortho-borylation of phenols, arylamines and alkylarenes. J Am Chem Soc. 2008; 130(24): 7534-7535. doi: 10.1021/ja8015878
- Kawamorita S, Ohmiya H, Hara K, Fukuoka A, Sawamura M. Directed ortho borylation of functionalized arenes catalyzed by a silica-supported compact phosphine-iridium system. J Am Chem Soc. 2009; 131(14): 5058-5059. doi: 10.1021/ja9008419
- Kawamorita S, Miyazaki T, Ohmiya H, Iwai T, Sawamura M. Rh-Catalyed ortho-selective C-H borylation of N-functionalized arenes with silicasupported bridge head monophosphine ligands. J Am Chem Soc. 2011; 133(48): 19310-19313. doi: 10.1021/ja208364a
- Ghaffari B, Preshlock SM, Plattner DL, et al. Silyl phosphorus and nitrogen donor chelates for homogeneous ortho-borylation catalysis. J Am Chem Soc. 2014; 136(41): 14345-14348. doi: 10.1021/ja506229s
- Bisht R, Chattopadhyay B. Formal Ir-catalyzed ligand-enabled ortho and meta borylation of aromatic aldehydes via in situ-generated imines. J Am Chem Soc. 2016; 138(1): 84-87. doi: 10.1021/jacs.5b11683
- Wang G, Liu L, Wang H, et al. N-B-Bidentate boryl ligand-supported iridium catalyst for efficient functional-group-directed C-H borylation. J Am Chem Soc. 2017; 139(1): 91-94. doi: 10.1021/jacs.6b11867
- Preshlock S, Plattner D, Maligres P, Maleczka R, Smith III MR. A traceless directing group for C-H borylation. Angew Chem Int Ed Engl. 2013; 52(49): 12915-12919. doi: 10.1002/anie.201306511
- Kuninobu Y, Ohmura T, Iwanaga T, Takai K. Palladium-catalyzed orthoselective C-H borylation of 2-phenylpyridine and its derivatives at room temperature. Angew Chem Int Ed Engl. 2013; 52(16): 4431-4434. doi: 10.1002/anie.201210328
- Li HL, Kuninobu Y, Kanai M. Lewis acid-base interaction-controlled orthoselective C-H borylation of aryl sulfides. Angew Chem Int Ed Engl. 2017; 56(6): 1495-1499. doi: 10.1002/anie.201610041
- Chattopadhyay B, Dannat JE, Andujar-De Sanctis IL, et al. Ir-Catalyzed ortho-borylation of phenols directed by substrate-ligand electrostatic interactions: A combined experimental/in silico strategy for optimizing weak interaction. J Am Chem Soc. 2017; 139(23): 7864-7871. doi: 10.1021/jacs.7b02232
- Kuninobu Y, Ida H, Nishi M, Kanai M. A meta-selective C-H borylation directed by a secondary interaction between ligand and substrate. Nature Chem. 2015; 7(9): 712-717. doi: 10.1038/nchem.2322
- Davis HJ, Mihai MT, Phipps RJ. Ion pair-directed regiocontrol in transitionmetal catalysis: a meta-selective C-H borylation of aromatic quaternary ammonium salts. J Am Chem Soc. 2016; 138(39): 12759-12762. doi: 10.1021/jacs.6b08164
- Saito Y, Segawa Y, Itami K. para-C-H borylation of benzene derivatives by a bulky iridium catalyst. J Am Chem Soc. 2015; 137(15): 5193-5198. doi: 10.1021/jacs.5b02052
- Yang LC, Semba K, Nakao Y. para-Selective C-H borylation of (Hetero) arenes by cooperative iridium/aluminum catalysis. Angew Chem Int Ed Engl. 2017; 56(17): 4853-4857.
- Hoque EM, Bisht R, Haldar C, Chattopadhyay B. Non covalent interactions Ir-catalyzed C-H activation: L-shaped ligand for para-selective borylation of aromatic esters. J Am Chem Soc. 2017; 139(23): 7745-7748.
