

Research Article
Quantum Chemical Study of Molecular Recognition between Etravirine/Rilpivirine and the HIV-1 Reverse Transcriptase
Department of Chemistry and Biochemistry, University of Toledo, Toledo, USA
*Corresponding author: Xiche Hu, Department of Chemistry and Biochemistry, University of Toledo, Toledo, USA, Email: xiche.hu@utoledo.edu
Received: November 6, 2017 Accepted: November 22, 2017 Published: November 28, 2017
Citation: Alqahtani S, Yang H, Hu X. Quantum Chemical Study of Molecular Recognition between Etravirine/Rilpivirine and the HIV-1 Reverse Transcriptase. Madridge J Nov Drug Res. 2017; 1(1): 25-34. doi: 10.18689/mjndr-1000105
Copyright: © 2017 The Author(s). This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Second generation non-nucleoside reverse transcriptase inhibitors (NNRTIs) etravirine and rilpivirine are essential components in the highly active antiretroviral therapy for the treatment of patients infected with human immunodeficiency virus type 1 (HIV-1). They are highly potent drugs against wild-type viruses and have exhibited excellent antiviral activities against some NNRTIs-resistant HIV-1 variants. In order to understand the underlying mechanism behind their robust resistance profile in comparison with the first generation NNRTIs nevirapine and efavirenz, it is necessary to quantitatively analyze their binding pockets in the wild-type HIV-1 reverse transcriptase (RT) and various HIV-1 RT mutants at the molecular level. Therefore, a high-level ab initio quantum chemical analysis was performed to decipher the molecular determinants for recognition of etravirine and rilpivirine by the wild-type RT and some RT mutants (K103N, K103N/Y181C, and K103N/L100I) of clinically important virus strains. Pair wise intermolecular interaction analysis determined the contribution of individual intermolecular interactions to the binding affinities between the second generation NNRTIs (etravirine or rilpivirine) and several variants of RTs, including the wild-type RT, and clinically relevant K103N, K103N/Y181C, and K103N/L100I mutant RTs. This quantitative analysis led to the identification of drug-protein interactions that persist despite mutations as well as to the evaluation of stabilization energy losses upon mutations. The results of this study enhanced our understanding of the molecular level mechanisms by which the second generation NNRTI drugs maintain their strong binding to mutant RTs. It is hoped that findings of this work would have a direct impact on designing new NNRTIs that are even more resilient to mutations in future.
Keywords: HIV-1 Reverse Transcriptase Inhibitors; Etravirine; Rilpivirine; Intermolecular Interactions, Quantum Chemical Calculations
Introduction
Human immunodeficiency virus type 1 reverse transcriptase (HIV-1 RT) represents an attractive target for the development of anti-AIDS therapies. This enzyme plays a pivotal role in the HIV replication cycle by converting single-stranded HIV RNA genome into double-stranded HIV-1 DNA that is subsequently integrated into the host cell's chromosome [1,2]. The current inhibitors of HIV-1 RT can be categorized into two main classes: nucleoside reverse transcriptase inhibitors (NRTIs) and non-nucleoside reverse transcriptase inhibitors (NNRTIs). The NRTIs are analogous of normal nucleotides but they lack the 3' hydroxyl group. They are competitive substrate inhibitors that incorporate into the nascent viral DNA and block the addition of new nucleotides, leading to the termination of DNA polymerization [3,4]. A major problem pertaining to the use of NRTIs is that they can also interfere with the DNA synthesis of the host cell, causing serious side effects [5,6]. NNRTIs, on the other hand, are highly specific non-competitive inhibitors of the HIV-1 RT that stop the retroviral enzyme replication and do not inhibit the host DNA polymerases [7,8,9]. By virtue of their selectivity, potency and low toxicity to the host, they are fundamental components in the highly active antiretroviral therapy (HAART) for the treatment of HIV-1 infection [10].
Despite the structural diversity of NNRTIs, they all bind to a common hydrophobic binding site in the RT enzyme referred to as the non-nucleoside inhibitor binding pocket (NNIBP), which is located at a distance of about 10 Å from the enzyme's polymerase active site [11,12]. Upon the binding of NNRTIs to their allosteric binding sites, an induced conformational change occurs at the enzyme's polymerase active site, rendering the enzyme catalytically inactive, thereby stopping DNA replication [7]. First generation NNRTIs, Nevirapine [13], Delavirdine [14], and Efavirenz [15] are drugs with a low genetic barrier that requires a single point mutation to trigger a high level resistance that severely impairs their binding affinities to RT [16,17]. In contrast, second generation NNRTIs Etravirine [18] and Rilpivirine [19] are potent inhibitors with a higher genetic barrier to resistance which allows them to retain potency against HIV-1 mutant strains that show resistance to first generation NNRTIs [17,20].
Like the vast majority of drugs, NNRTIs bind to HIV-1 RT via non covalent interactions (e.g. hydrogen bonding, π-π stacking interactions, CH-π interactions and cation-π interactions). The binding pose of an inhibitor into its receptor, is determined by the collection of all the attractive and repulsive forces between the inhibitor and its surrounding interaction partners. Therefore, to gain an in-depth insight of the molecular recognition between a NNRTI and its receptor, it is of great importance to quantify the specific contributions of each amino acid in the binding pocket to the binding affinity between the NNRTI and its receptor. Deciphering the energetic contribution of the individual residues in the ligand binding pocket toward protein-NNRTI binding would have a great influence on designing new inhibitors. Binding affinity data determined experimentally represents the collective contributions of all protein residues that form the ligand binding pocket. Quantum chemical calculations, on the other hand, offer a way to rigorously quantify the magnitude and characterize the nature of individual intermolecular interactions between ligands and their surrounding protein residues.
The aim of the present study is to determine the underlying reason behind the origin of high binding affinities of second generation NNRTIs etravirine and rilpivirine to the wild-type HIV-1 RT and some clinically relevant mutant variants of the virus at the molecular level. For this purpose, data mining and high level quantum chemical calculations were performed to investigate the molecular recognition between etravirine/ rilpivirine and wild-type RT and mutated RTs. The energetic contributions of the individual intermolecular interaction between etravirine or rilpivirine and their surrounding amino acids to the binding affinities between these inhibitors and the various RTs were systematically analyzed in a pair wise manner. The results of this study will not only shed light on the molecular determinants for the molecular recognition of NNRTIs in HIV-1 RT, but also enhance our understanding of molecular recognition of drugs in protein in general. Therefore, this work is expected to have a direct impact on designing the next generation of NNRTIs with a higher potency and even more resilience to mutations
Methods and Theory
Data Mining
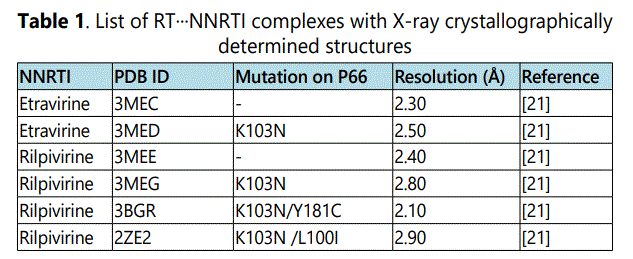
The availability of crystal structures of etravirine and rilpivirine bound to the wild-type HIV-1 RT and some clinically important single and double point mutants from Protein Data Bank (PDB) forms the basis of our quantum chemical analysis present here. Table 1 lists the six PDB files of NNRTIs (etravirine and rilpivirine) bound to RTs that were selected and analyzed in this study.
For all the calculations considered in this study, the atomic coordinates of non-hydrogen atoms in NNRTIs (etravirine and rilpivirine) and their interacting protein residues were extracted from the X-ray crystal structures of the complexes. The missing hydrogen atoms were added and their positions were determined by ab initio geometry optimization at the HF/6-31+G* level. During the geometry optimization processes, the positions of heavy (non-hydrogen) atoms were fixed. All geometry optimization calculations were carried out using the Gaussian09 software [23].
Intermolecular Interaction Energies in the Gas Phase
For a given level of theoretical method, the quality of quantum mechanical calculation depends on the level of theory used and the choice of the basis set. For an accurate description of the intermolecular interactions between ligands and their surrounding protein residues, the electron correlation must be taken into consideration. Intermolecular interactions between neutral molecules are usually dominated by dispersion interactions [24,25] which is caused by the mutual correlation of electrons that belong to the interacting partners. The magnitude of correlation energy can be as high as the magnitude of the intermolecular interaction itself. Therefore, the inclusion of electron correlation is of a great importance for a proper description of non-bonded interactions in biological systems. The M∅ller–Plesset perturbation theory at the second order (MP2) represents a feasible and popular method that recovers a significant portion of electron correlations [26]. In our study, pair wise intermolecular interaction energies, in the gas phase, between etravirine or rilpivirine and their surrounding protein residues were calculated by ab initio electronic structure calculations at the MP2 level combined with the triple-zeta cc-pVTZ Dunning's correlation basis set for an adequate treatment of electron correlation.
The interaction energies between inhibitors and their interacting protein partners were estimated at the MP2/cc-pVTZ level using the super molecular approach in which the interaction energy between the two molecules (e.g. A and B) is computed as the potential energy difference between the energy of the interacting dimer EAB and the energies of the monomers EA and EB:
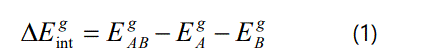
The basis set superposition error (BSSE) was corrected by the Boys and Bernardi Counter Poise Method [27]. All calculations were carried out using the Gaussian09 software [23].
Free Energies of Solvation
Since all biological processes take place in an aqueous environment in living organisms, the solvation effect has to be taken into account in order to obtain a reliable estimate of binding strengths of the intermolecular interactions in biological systems. A number of different solvation models have been developed to deal with this issue. Depending on the way solvent molecules are treated, these models can be divided into two main classes: implicit solvation models and explicit solvent models [28,29]. In explicit solvation models, all of the interactions in the system are fully described at the atomic level, which means that all the solvent molecules are explicitly incorporated in quantum mechanics or molecular mechanics calculations. Unfortunately, explicit treatment of solvent molecules when simulating biological systems at the quantum chemical level is not feasible due to the prohibitively high computational cost. Instead, a common way to calculate the free energy of solvation when dealing with biological systems is using the implicit solvation models. In these models, the solvent is described as a continuum, a polarizable medium with a fixed dielectric constant equals to the bulk value for pure solvent. Here, we employed the implicit solvation model SM5.42R developed by Truhlar and his coworkers [30,31] at the HF/6-31+G* level as implemented in the GAMESS-version 11 Apr. 2008 (R1) software package [32] to quantify the free energy of solvation ΔGsol of the different species considered in this study.
The standard-state free energy of solvation ΔGsol in the SM5.42R model is composed of two components: the electrostatic term and the first-solvation-shell (nonelectrostatic) term. When the solute is solvated, the charge distribution of the solute polarizes the surrounding homogenous dielectric medium of solvent and subsequently, the polarized field of the solvent acts back on the solute electric charge distribution to be self-consistent with the solvent electric polarization. The electrostatic term includes solute-solvent favorable interactions, solvent rearrangement cost as well as the energy cost resulting from the distortion of solute charge distribution. The first-solvation-shell term includes contributions arising from cavity creation, dispersion and change in the solvent structure. Before the protein-NNRTI complex is formed in solution, both the protein and NNRTI are solvated and have to lose part of their solvation shells upon binding which incurs an energy cost commonly referred to as dehydration energy. For a general reaction of A combines with B to form the AB complex, the dehydration energy for the complex formation is given by the following equation:
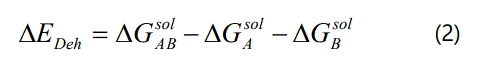
where, ΔGisol = AB, A, B represents the free energies of solvation for the complex AB, and the monomers A, B, respectively.
Intermolecular Interaction Energies in Solution
The binding energy for complex formation in solution was indirectly obtained as a sum of intermolecular interaction energies in the gas phase (Equation 1) and the dehydration energies for the complex formation in solution (Equation. 2) as follows:
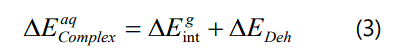
Results and Discussion
Since the main objective of this study is to identify molecular determinants for molecular recognition of second generation NNRTIs etravirine and rilpivirine in the wild-type RT and its mutants, we carefully examined crystal structures of etravirine or rilpivirine bound to the wild-type HIV-1 RT and mutant strains that are listed in Table 1. Despite the extensive experimental efforts on studying the mechanism behind the persistent potency of the secondary generation NNRTIs etravirine and rilpivirine against wild-type RT and mutated viruses, quantum chemical analysis is a very useful tool to quantify the contributions of the individual amino acids in the non-nucleoside inhibitor binding pocket (NNIBP) to the overall binding affinities between these drugs and RTs. The binding environments of both drugs in the wild-type RT and mutant strains were systematically examined using the Visual Molecular Dynamic (VMD) program [36] that is capable of three-dimensional stereographic display. Based on the 3D analysis of the crystal structures of RT…NNRTI complexes, amino acids residues of RTs that form the binding pockets of etravirine and rilpivirine were systematically identified. Subsequently, gas phase intermolecular interaction energies between these inhibitors and their interacting amino acid residues were calculated by means of the super molecular approach at the MP2/cc-pVTZ level, followed by solvation energy correction based on the implicit solvation model SM5.42R at the HF/6-31+G* level.
Binding of Etravirine/Rilpivirine to the Wild-Type HIV-1 RT
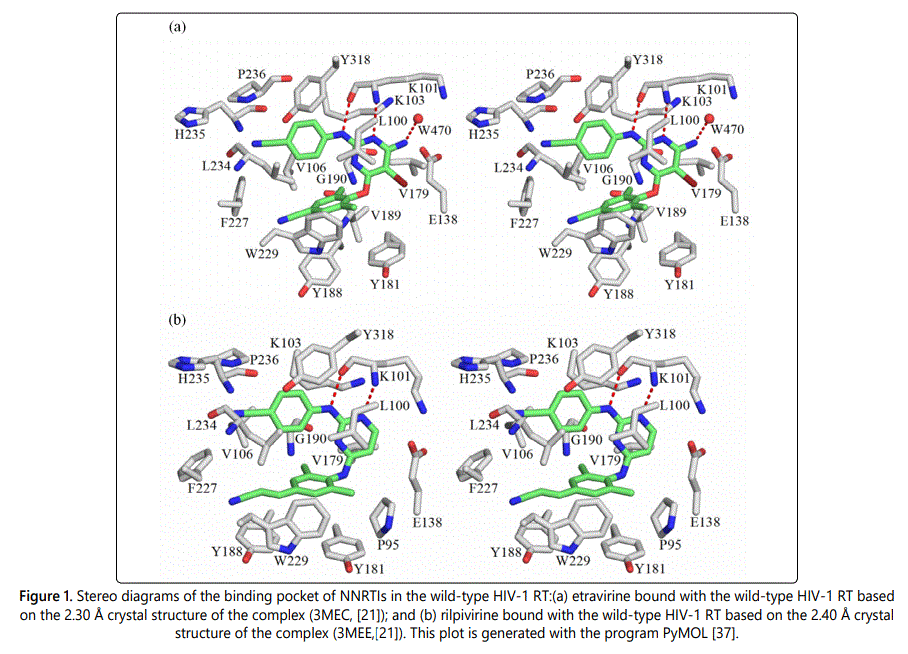
Due to the similar structural frames of the second generation NNRTIs etravirine and rilpivirine, both inhibitors adopt, to a large extent, very similar binding modes to RTs. Their interacting protein residues are located in the p66 subunit with the exception of Glu138 which belongs to the p51 subunit. The following amino acids Gly190, Val106, Val179, Leu100, Leu234, Pro236, Phe227, Tyr181, Tyr188, Tyr318, Trp229, His235, Lys101, Lys103, and Glu138 are located within 4.8 Å of both inhibitors and have the potential, based on 3-D analysis of structures, to be involved in favorable interactions with the inhibitors. The only two noticeable differences are that, etravirine seems to have two additional favorable interactions with Val189 and a water molecule (residue number 470 in the p51 subunit). The latter appears to bridge etravirine to RT enzyme via a mediated hydrogen bond. In addition, rilpivirine appears to be interacting with Pro95.
Figure 1 shows stereographic displays of the binding pockets of etravirine and rilpivirine in the wild-type HIV-1 RT. Hydrogen bonds are shown as red dotted lines. Based on the above identification of intermolecular contacts, a high level quantum chemical analysis was employed to quantify the magnitude of intermolecular interaction energies between etravirine and rilpivirine and their interacting protein residues. Listed in Table 2 and Table 3 are the pair wise intermolecular interaction energies between etravirine/rilpivirine and the protein residues calculated at the MP2/cc-pVTZ level in the gas phase (ΔEMP2) and after solvation correction (ΔEsol). Intermolecular interaction energy calculations after taking solute-solvent interaction into account yields the complete picture of interactions in a cellular environment. Therefore, the interaction energies after solvation correction presented in this study represents the complete picture of the binding strength of intermolecular interactions between these drugs and their interacting residues of RTs. The specific type of non covalent intermolecular interactions between etravirine/ rilpivirine and amino acids of RT are identified and shown in parentheses.
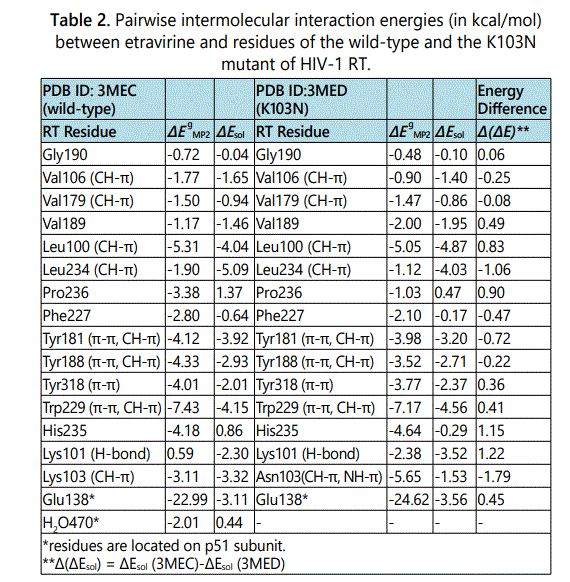
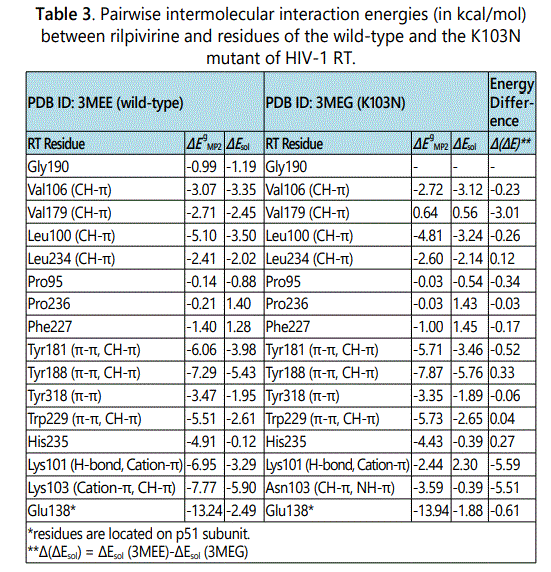
In the case of binding of etravirine to the wild-type HIV-1 RT, as shown in Table 2, all of the gas phase interaction energies between etravirine and the residues of RT are attractive (negative) with the exception of the interaction energy between etravirine and Lys101 which produced a repulsive energy (positive). After solvation correction, intermolecular interaction energies between etravirine and its binding pocket residues in RT are attractive as indicated by their negative values. An exception is observed in the case of the interactions with Pro236 and the water molecules (residue number 470, p51), which give rise to repulsive interactions (positive values) after solvation correction. The overall binding energy between etravirine and the wild-type HIV-1 RT as calculated in solution and estimated as the total of all the favorable intermolecular interaction energies between etravirine and its binding pocket residue in RT is -35.60 kcal/mol. In the case of binding of rilpivirine to the wild-type HIV-1 RT, as shown in Table 3, all of the gas phase interaction energies between rilpivirine and the residues of RT are attractive (negative). After solvation correction, the intermolecular interaction energies between rilpivirine and its interacting amino acids of RT remain attractive except with Phe227 and Pro236 which interact unfavorably as implied by the positive values of their intermolecular interaction energies with rilpivirine. The overall binding energy for the binding of rilpivirine to the wild-type HIV-1 RT predicted in solution and calculated as the sum of all the attractive interactions between rilpivirine and its binding pocket residues is -39.16 kcal/mol.
As stated in the method section, prior to complex formation, both the HIV-1 RT and NNRTI are solvated and as they approach each other both molecules lose parts of their solvation shell upon binding to each other, which incurs an energy cost. Therefore, the interactions energies after solvation correction is usually lower than gas phase interaction energies especially for hydrogen bonding interactions, electrostatic interactions (salt-bridge) and cation-π interactions. The strengths of hydrophobic types of interactions such as π-π stacking interactions and CH-π interactions are less affected by solvation correction. As shown in Table 2 and Table 3, the magnitudes of the intermolecular interaction energies between etravirine/rilpivirine and RT after solvation correction are substantially large. This, to a large extent, can be attributed to the substantial contributions from π-π interactions and CH-π interactions between etravirine or rilpivirine and the aromatic and aliphatic residues of RT. Lys101 forms strong hydrogen bonding interactions with both inhibitors. Lys103 interacts strongly with etravirine and rilpivirine in the wild-type RT via CH-π and appears to be also forming cation-π interaction with rilpivirine.
Binding of Etravirine/Rilpivirine to the K103 Mutant
The K103N mutation is the most frequent mutation observed in viruses isolated from patients treated with NNRTIs [38,39]. It has been found that the K103N single point mutation is sufficient to confer high level of resistance to the first generation NNRTIs evirapine and efavirenz, reducing their susceptibility by 46 and 19-fold, respectively [40,41]. The second-generation NNRTIs etravirine and rilpivirine, on the other hand, show good profiles of activity against the single point K103N mutant, which allow them to retain potency against this mutant virus [42,43]. To understand the molecular basis of high binding affinities between etravirine/rilpivirine and K103N RT, we have quantitatively analyzed binding pockets of both drugs in the K103N RT mutant.
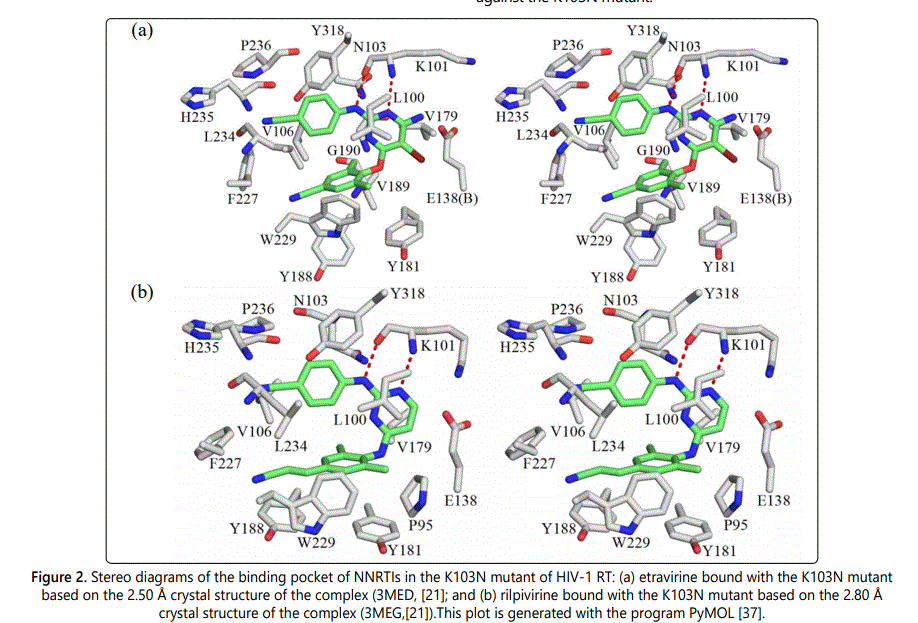
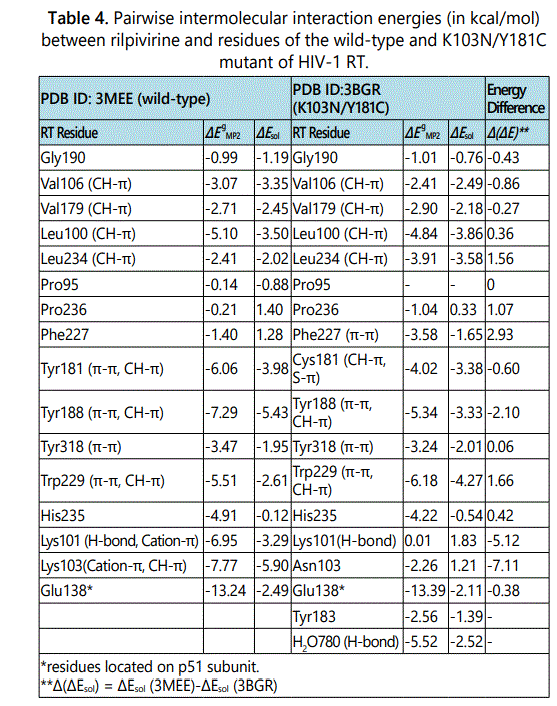
Following the same procedure as before, the intermolecular interactions between etravirine/rilpivirine and residues of K103N RT were energetically analyzed. Figure 2 shows the stereographic displays of etravirine and rilpivirine binding pockets in the K103N mutant. Table 2 and Table 3 list the resulting intermolecular interaction energies in the gas phase (ΔEMP2) and after solvation correction (ΔEsol) between etravirine and rilpivirine and residues of K103N mutant. Table 2 also presents a pair wise comparison of the intermolecular interaction energies between etravirine and residues of K103N mutant RT with those between etravirine and the wild-type RT residues. It can be seen that most residues in the K103N RT mutant maintain strong intermolecular interaction energies with etravirine in magnitudes that are roughly comparable to their binding strength with etravirine in the with wild-type RT. However, mutation of Lys103 to an asparagine results in a reduction of the solution phase interaction energies between etravirine and residue #103 from -3.32 kcal/molto -1.53 kcal/mol. Although the hydrogen bond interaction between etravirine and Lys101 is lost upon the K103N mutation, etravirine interacts favorably with Asn103 via NH-π and CH-π interactions. This single point mutation does not have any other noticeable effect on the intermolecular interactions between etravirine and the remaining amino acids in the binding pocket of etravirine. The favorable intermolecular interactions between etravirine and its binding pocket residues in the K103N mutant add up to a total of -35.12 kcal/mol stabilization energy. This strong binding affinity between etravirine and the K103N mutant is comparable to the binding affinity between etravirine and the wild-type RT.
Intermolecular interaction energies between rilpivirine and its binding pocket residues in the K103N mutant of HIV-1 RT are shown in Table 3, in comparison with those between rilpivirine and the wild-type RT residues. Mutation of lysine residue at position 103 to an asparagine resulted in a reduction of interaction energies from -5.90 kcal/mol to -0.39 kcal/mol, which represents a stabilization energy loss of -5.51 kcal/mol. Additionally, there are two significant stabilization energy losses at residue Lys101 (-5.59 kcal/mol stabilization energy loss) and at residue Val179 (-3.01 kcal/mol stabilization energy loss). Based on a careful examination of the optimized structures, it appears that Lys101 in wild-type RT interacts with rilpivirine via two hydrogen bonds and strong cation-π interactions meanwhile there is only one hydrogen bond formed between the carbonyl group of Lys101 and its adjacent NH group of rilpivirine in the K103N mutant of RT. In addition, there is change in the position of amino group of the Lys101 residue upon K103N mutation, which might have weakened cation-π interactions between Lys101 and rilpivirine. It was also observed that, the side chain of Val179 change its position with respect to the rilpivirin ephenyl ring nearby. One of the CH3 groups of Val179 was pointing to the centroid of the rilpivirine aromatic ring in the wild-type RT, which results in strong CH-π interaction. Upon K103N mutation, the side chain of Val179 rotates so that CH3 is no longer pointing toward the centroid of the aromatic ring of rilpivirine nearby. However, despite these stabilization energy losses, the remaining residues of the rilpivirine binding pocket in K103N RT maintain strong intermolecular interactions with rilpivirine. The total stabilization energy between rilpivirine and K103N RT residues is -25.46 kcal/mol. Despite K103N mutation, both etravirine and rilpivirine maintain strong interactions with a large number of RT residues, which enables these inhibitors to overcome drug resistance and retain potency against the K103N mutant.
Binding of Rilpivirine to the K103N/Y181C double Mutant
The double drug resistance mutations K103N and Y181C are among the most commonly observed mutations in patients treated with NNRTIs. The double mutation K103N/Y181C severely impaired the effectiveness of the first generation NNRTIs nevirapine and efavirenz. It has been found that K103N/Y181C double-point mutation reduces nevirapine and efavirenz potencies by 625 and 1176 fold, respectively [22]. Second generation NNRTIs etravirine and rilpivirine potently inhibit K103N/Y181C double mutant with EC50 values less than one nanomolar [44]. Only the X-ray crystal structure of the K103N/Y181C RT double mutant bound to rilpivirine was available from the Protein Data Bank (PDB). Therefore, we were able to conduct a quantitative analysis of the intermolecular interactions between rilpivirine and its binding pocket residues in the K103N/Y181C RT double mutant. Following the same computational procedure as described earlier, intermolecular interactions between rilpivirine and the residues of K103N/Y181C RT were systematically analyzed. Figure 3 shows the stereographic drawing of the rilpivirine binding pocket in the K103N/Y181C mutant. Potential hydrogen bonds are shown as red dotted lines.
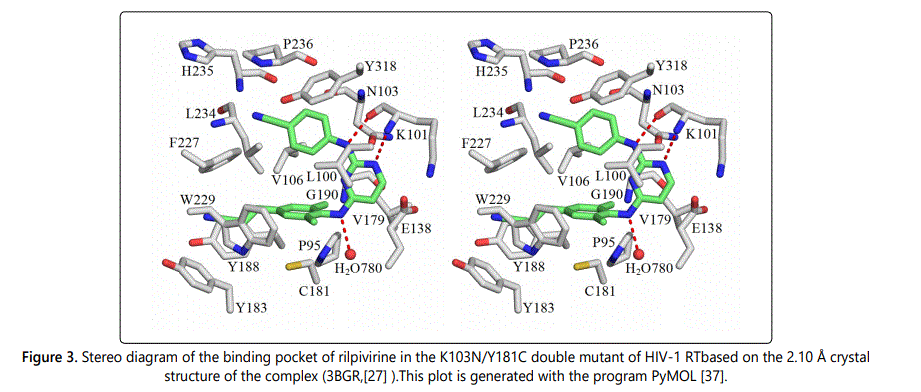
Listed in Table 4 are pair wise intermolecular interaction energies in gas phase (ΔEMP2) and after solvation correction (ΔEsol) between rilpivirine and its interacting residues in the K103N/Y181C double mutant of HIV-1 RT. For the purpose of comparison, the interaction energies between rilpivirine and amino acids of the wild-type are also listed in Table 4. Comparing the intermolecular interaction energies between rilpivirine and the K103N/Y181C mutant with those between rilpivirine and the wild-type RT, we can see that the doublepoint mutation K103N/Y181C severely impaired the intermolecular interactions between rilpivirine and enzyme residues at position 101 and 103, in a similar manner as in the case of the K103N mutant. The pair wise intermolecular interaction energy, in solution, between rilpivirine and Lys101 changes from a favorable -3.29 kcal/mol in wild-type RT to an unfavorable 1.83 kcal/mol in the K103N/Y181C mutant. This is a -5.12 kcal/mol stabilization energy loss that can be attributed to the loss of hydrogen bonding interaction and cation-π interaction at residue 101 as a result of the K103N/Y181C mutation. Another great loss of intermolecular interaction energy is found at residue 103. Upon mutation of Lys103 to an asparagine, the interaction energy, in solution, changes from a favorable -5.90 kcal/mol for rilpivirine-Lys103 (in wildtype) to an unfavorable 1.21 kcal/mol for rilpivirine-Asn103 in the double mutant RT. This is a -7.11 kcal/mol stabilization energy loss that can be attributed to the loss of cation-π, CH-π interactions at residue 103 as a result of the K103N/ Y181C mutations. Additionally, it was found that Asn103 forms a repulsive interaction with rilpivirine after solvation correction. By examining intermolecular interactions between rilpivirine and the other residues in the binding pocket, we found that rilpivirin retains strong interactions with residue 181 after the Y181C mutation. Some other points worth mentioning here are that, interaction energy after solvation correction for the rilpivirine-Phe227 pair changes from an unfavorable 1.28 kcal/molto a favorable -1.65 kcal/mol as a result of the K103N/Y181C mutation. In addition, there is an enhancement of interaction energies at residue W229. As can be seen in Table 4, the interaction energy between rilpivirine and W229 changes from -2.61 kcal/mol to -4.27 kcal/mol upon the double mutation. It has been also found that the binding of rilpivirine to the K103N/Y181C mutant is enhanced by forming two additional attractive interactions with residues Tyr183 and a water molecule (residue number 780 in the p51 subunit). The total stabilization energies gained from these two new interactions are -1.39 and -2.52 kcal/mol, respectively. The total stabilization energy for the formation of drugprotein complex between rilpivirine and the K103N/Y181C double mutant, calculated as the summation of favorable intermolecular interactions after solvation correction, is -34.07 kcal/mol.
Binding of Rilpivirine to the K103N/L100I double Mutant
The K103N/L100I double mutation is another common combination of mutations that has a severe impact on the effectiveness of the first generation NNRTIs nevirapine and efavirenz. Although, the K103N/L100I double-point mutation has a larger effect on the potency of rilpivirine than K103N and K103N/Y181C mutations, it has been found that second generation NNRTI rilpivirine retains excellent activity against the K103N/L100I double mutant virus with an EC50 value of ~ 8nM [22]. The availability of X-ray structure of rilpivirine bound to the K103N/L100I mutant of HIV-1 RT enabled us to quantify the magnitude of intermolecular interaction energies between rilpivirine and its binding pocket residues in the K103N/L100I double mutant. Following a similar manner for analyzing drug-protein interactions, the intermolecular interactions between rilpivirine and residues of K103N/L100I HIV-1 RT were systematically analyzed. Figure 4 shows a stereographic drawing of the rilpivirine binding pocket in the K103N/L100I mutant. Hydrogen bond between rilpivirine and Lys101 is shown as red dotted line.
Table 5 lists the intermolecular interaction energies in gas phase (ΔEMP2) and after solvation correction (ΔEsol) between rilpivirine and its interacting residues in the K103N/L100I mutant. For an easy comparison of intermolecular interaction energies between rilpivirine and the K103N/L100I double mutant residues with those between rilpivirine and residues of the wild-type RT, the intermolecular interaction energies between rilpivirine and its binding pockets residues in both RTs are listed side by side. It was found that the attractive interaction between rilpivirine and Lys101 via hydrogen and cation-π interaction is lost upon the K103N/L100I mutation. After solvation correction the intermolecular interaction energy between rilpivirine and Lys101 changes from a favorable -3.29 kcal/mol (attraction) to an unfavorable 2.38 kcal/mol (repulsion).
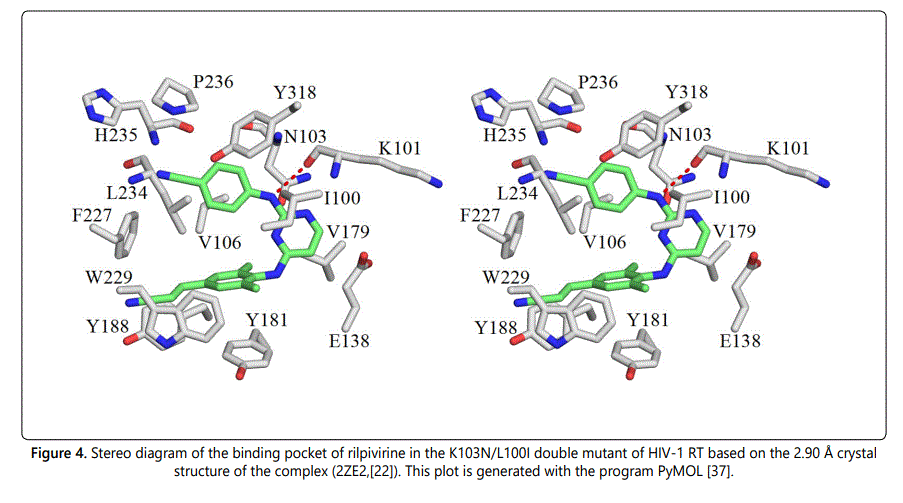
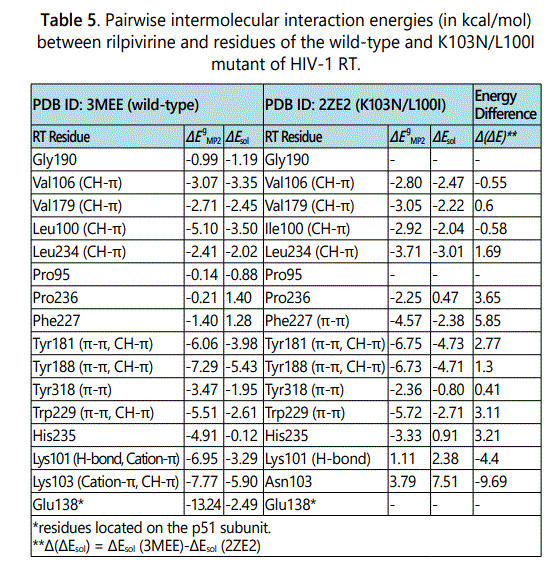
Another significant stabilization energy loss occurred at residue 103. The interaction energy changes from a favorable -3.29 kcal/mol between rilpivirine and Lys103 to an unfavorable 7.51 kcal/mol between rilpivirine and Asn103. This can be attributed to the loss of cation-π and CH-π interactions at residue 103 as a consequence of the K103N/L100I double mutation. A new strong favorable interaction is formed between rilpivirine and Phe227 in the K103N/L100I mutant. Mutation at residue 100 in which the leucine residue is replaced by an isoleucine has a subtle effect on the magnitude of the interaction energy between rilpivirine and residue 100. Both Leu100 in the wild-type and Ile100 in the K103N/L100I mutant form strong CH-π interactions with rilpivirine. The magnitudes of these intermolecular interaction energies, in solution, are-3.50 kcal/ mol and -2.04 kcal/mol, respectively. Most of the remaining amino acids in the binding pocket of rilpivirine in the K103N/ L100I mutant form favorable interactions that are comparable to those between rilpivirine and wild-type RT residues. The total stabilization energy between rilpivirine and residues of the K103N/L100I double mutant RT add up to -25.07 kcal/mol. This strong stabilization energy explains the good activity of rilpivirine against the K103N/L100I mutant of RT.
Conclusion
In summary, a high level quantum chemical analysis was carried out to analyze molecular determinants for recognition between the second generation NNRTIs etravirine and rilpivirine in HIV-1 RTs. To understand the origin of the potency of these drugs against wild-type and mutant RTs, the contributions of the individual amino acid residues in the NNRTI-binding pocket to the overall binding forces between etravirine/rilpivirine and both the wild-type RT and some clinically important mutants RTs were quantum chemically evaluated. In addition to the large flexibility of these drugs that allows them to adopt different conformations in response to the different mutations, non-bonded interaction involving the aromatic moieties of these drugs are critical for their excellent activity against the wild-type RT and its mutant strains. The pair wise intermolecular interaction calculations conducted in our study have deciphered the molecular level drug-protein interactions that help these drugs to overcome drug resistance. It has been discovered that, unlike the first generation of NNRTIs nevirapine and efavirenz that rely heavily for their binding to RT on their interactions with Tyr181 and Tyr188, both etravirine and rilpivirine attain their tight binding to RT and its mutants by forming strong favorable interactions with a wide range of their binding pockets residues including the highly conserved residues Trp229, Leu234, Phe227 and Tyr318. Therefore, any stabilization energy losses resulting from single or double point mutations of the non-nucleoside inhibitor binding pocket (NNIBP) have less effects on etravirine and rilpivirine potencies. The results of our quantum chemical calculations presented in this study, enhanced our understanding of molecular recognition between the second generations NNRTI etravirine/rilpivirine and RTs, unravelling at the molecular level the mechanisms that used by these drugs to achieve their high binding affinities to the clinically relevant mutants of RTs. It is hoped that the insights gained thorough this rigorous comparative theoretical investigation of molecular determinants for binding of NNRTIs with their targeted wild type and mutant RTs will lay the foundation for designing the next generation of NNRTIs that are even more resilient to mutations.
Acknowledgment
It is our great pleasure to acknowledge the Ohio Supercomputer Center for a generous allocation of supercomputer time.
References
- Katz RA, Skalka AM. The Retroviral Enzymes. Annual Review of Biochemistry. 1994; 63(1): 133-173. doi: 10.1146/annurev.bi.63.070194.001025
- Sluis-Cremer N, Arion D,Parniak MA. Molecular mechanisms of HIV-1 resistance to nucleoside reverse transcriptase inhibitors (NRTIs). Cell Mol Life Sci.2000; 57(10): 1408-1422. doi: 10.1007/PL00000626
- Hartman TL, Buckheit RW. The Continuing Evolution of HIV-1 Therapy: Identification and Development of Novel Antiretroviral Agents Targeting Viral and Cellular Targets. Molecular Biology International. 2012. doi: 10.1155/2012/401965
- De Clercq E, The role of non-nucleoside reverse transcriptase inhibitors (NNRTIs) in the therapy of HIV-1 infection. Antivir Res.1998; 38(3): 153-179. doi: 10.1016/S0166-3542(98)00025-4
- de Bethune MP. Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in the treatment of HIV-1 infection: A review of the last 20 years (1989-2009). AntivirRes. 2010; 85(1): 75-90. doi: 10.1016/j.antiviral.2009.09.008
- Ren J, Esnouf R, Garman E, Somers D, Ross C,Kirby I, et al. High resolution structures of HIV-1 RT from four RT-inhibitor complexes. Nat Struct Mol Biol. 1995; 2(4): 293-302. doi: 10.1038/nsb0495-293
- Ren J, Milton J, Weaver KL, Short SA, Stuart DI, Stammers DK. Structural Basis for the Resilience of Efavirenz (DMP-266) to Drug Resistance Mutations in HIV-1 Reverse Transcriptase, Structure.2000; 8(10): 1089-1094. doi: 10.1016/S0969-2126(00)00513-X
- Hsiou Y, Ding JP, Das K, Clark AD, Boyer PL, et al. The Lys103Asn mutation of HIV-1 RT: A novel mechanism of drug resistance, J Mol Biol.2001; 309(2): 437-445. doi: 10.1006/jmbi.2001.4648
- Usach I, Melis V, Peris JE, Non-nucleoside reverse transcriptase inhibitors: a review on pharmacokinetics, pharmacodynamics, safety and tolerability. J Int Aids Soc.2013; 16(1). doi: 10.7448/IAS.16.1.18567
- Das K, Clark AD, Lewi PJ, Heeres J, de Jonge MR, et al. Roles of Conformational and Positional Adaptability in Structure-Based Design of TMC125-R165335 (Etravirine) and Related Non-nucleoside Reverse Transcriptase Inhibitors That Are Highly Potent and Effective against Wild-Type and Drug-Resistant HIV-1 Variants. J Med Chem. 2004; 47(10): 2550-2560. doi: 10.1021/jm030558s
- Janssen PAJ, Lewi PJ, Arnold E, Daeyaert F, de Jonge M, et al. In search of a novel anti-HIV drug: Multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1E)-2-cyanoethenyl]-2,6dimethylphenyl] amino]-2-pyrimidinyl] amino]-benzonitrile (R278474, rilpivirine). J Med Chem. 2005; 48(6): 1901- 1909. doi: 10.1021/jm040840e
- Lansdon EB, Brendza KM, Hung M, Wang R, Mukund S, et al. Crystal Structures of HIV-1 Reverse Transcriptase with Etravirine (TMC125) and Rilpivirine (TMC278): Implications for Drug Design. J Med Chem. 2010; 53(10): 4295-4299. doi: 10.1021/jm1002233
- Das K, Bauman JD, Clark AD, Frenkel YV, Lewi PJ, et al. High-resolution structures of HIV-1 reverse transcriptase/TMC278 complexes: Strategic flexibility explains potency against resistance mutations. P Natl Acad Sci USA. 2008; 105(5): 1466-1471. doi: 10.1073/pnas.0711209105
- Buckingham AD, Fowler PW, Hutson JM. Theoretical studies of van der Waals molecules and intermolecular forces. Chemical Reviews. 1988; 88(6) 963-988.
- Chalasinski G, Gutowski M. Weak interactions between small systems. Models for studying the nature of intermolecular forces and challenging problems for ab initio calculations. Chemical Reviews.1988; 88(6): 943-962. doi: 10.1021/cr00088a007
- Boys SF, Bernardi F. The calculation of small molecular interactions by the differences of separate total energies.Some procedures with reduced errors (Reprinted from Molecular Physics, vol 19, pg 553-566, 1970). Mol Phys. 2006; 100(1): 65-73. doi: 10.1080/00268977000101561
- Cramer CJ, Truhlar DG. Implicit solvation models: Equilibria, structure, spectra, and dynamics. Chemical Reviews. 1999; 99(8): 2161-2200. doi: 10.1021/cr960149m
- Tomasi J, Persico. Molecular-Interactions in Solution - an Overview of Methods Based on Continuous Distributions of the Solvent. Chemical Reviews.1994; 94(7): 2027-2094. doi: 10.1021/cr00031a013
- Li JB, Hawkins GD, Cramer CJ, Truhlar DG. Universal reaction field model based on ab initio Hartree-Fock theory, Chem Phys Lett. 1998; 288(2-4): 293-298. doi: 10.1016/S0009-2614(98)00194-8
- Zhu TH, Li JB, Hawkins GD, Cramer CJ, Truhlar DG, Density functional solvation model based on CM2 atomic charges. J Chem Phys. 1998; 109(20): 9117-9133. doi: 10.1063/1.476521
- Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon M.S,et al.Montgomery, General Atomic and Molecular Electronic-Structure System. J ComputChem. 1993; 14(11): 1347-1363. doi: 0.1002/jcc.540141112
- Mao LS, Wang YL, Liu YM, Hu XC. Multiple intermolecular interaction modes of positively charged residues with adenine in ATP-binding proteins. J Am Chem Soc. 2003; 125(47): 14216-14217. doi: 10.1021/ja036096p
- Liu YM, Hu XH. Molecular determinants for binding of ammonium ion in the ammonia transporter AmtB - A quantum chemical analysis. J Phys Chem A. 2006; 110(4): 1375-1381. doi: 10.1021/jp054261c
- Mao LS, Wang YL, Liu YM, Hu XC. Molecular determinants for ATP-binding in proteins: A data mining and quantum chemical analysis. J Mol Biol. 2004; 336(3): 787-807. doi: 10.1016/j.jmb.2003.12.056
- Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph Model. 1996; 14(1): 33-38. dio: 10.1016/0263-7855(96)00018-5
- Re MC, Monari P, Bon I, Borderi M, Gibellini D, Schiavone, et al. Development of drug resistance in HIV-1 patients receiving a combination of stavudine, lamivudine and efavirenz. Int J Antimicrob. 2002; 20(3): 223- 226. doi: 10.1016/S0924-8579(02)00164-4
- Bacheler L, Jeffrey S, Hanna G, D'Aquila R, Wallace L, et al. Genotypic correlates of phenotypic resistance to efavirenz in virus isolates from patients failing nonnucleoside reverse transcriptase inhibitor therapy. J Virol. 2001; 75(11): 4999-5008. doi: 10.1128/JVI.75.11.4999-5008.2001
- Rhee SY, Liu T, Ravela J, Gonzales MJ, Shafer RW. Distribution of human immunodeficiency virus type 1 protease and reverse transcriptase mutation patterns in 4,183 persons undergoing genotypic resistance testing. Antimicrob Agents.Ch. 2004; 48(8): 3122-3126. doi: 10.1128/ AAC.48.8.3122-3126.2004
- Andries K, Azijn H, Thielemans T, Ludovici D, Kukla M,et al. TMC125, a novel next-generation nonnucleoside reverse transcriptase inhibitor active against nonnucleoside reverse transcriptase inhibitor-resistant human immunodeficiency virus type 1. Antimicrob Agents Ch. 2004; 48(12): 4680-4686. doi: 10.1128/AAC.48.12.4680-4686.2004
- Azijn M, Tirry I, Vingerhoets J, de Bethune MP, Kraus G,et al. TMC278, a Next-Generation Nonnucleoside Reverse Transcriptase Inhibitor (NNRTI), Active against Wild-Type and NNRTI-Resistant HIV-1. Antimicrob Agents Ch. 2010; 54(2): 718-727. doi: 10.1128/AAC.00986-09
- Heeres J, Lewi PJ. The medicinal chemistry of the DATA and DAPY series of HIV-1 non-nucleoside reverse transcriptase inhibitors. Advances in Antiviral Drug Design. 2007; 213-242. doi: 10.1016/S1075-8593(06)05006-4
