

Research Article
Opto-Electronic Properties of Li2C2 Polymorphs
1Department of Physics, GC University Faisalabad, Faisalabad, Pakistan
2School of Mathematical and Physical Sciences, University of Technology Sydney, Australia
*Corresponding author: Sajid Ali, School of Mathematical and Physical Sciences, University of Technology Sydney, Australia, Tel: +61 449764974, E-mail: Sajid.Ali@student.uts.edu.au
Received: August 16, 2017 Accepted: August 28, 2017 Published: September 4, 2017
Citation: Ali S. Opto-Electronic Properties of Li2C2 Polymorphs. Madridge J Nanotechnol Nanosci. 2017; 2(1): 73-75. doi: 10.18689/mjnn-1000113
Copyright: © 2017 The Author(s). This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
The electronic and optical properties of Li2C2 in different structural phases are studied in the frame work of density functional theory. Full potential linearized augmented plane waves plus local orbitals (FP-LAPW+lo) method is applied within the Engel-Voskogeneralized gradient approximation. The compound is metallic in Bmmb phase, while insulating in Immm and I21 3 phases. The calculated bandgaps are in close agreement with the previous predictions. The two phases Immm I21 3 and show a high dielectric function and optical conductivity in the UV range and are therefore suitable to be used in the fabrication of optoelectronic devices.
Keywords: Optical Conductivity; Fabrication; Optoelectronic devices; Bandgaps.
Introduction
Metal-intercalated graphite Li2C2 owing to its unique physical and chemical properties have been of scientific [1-3] and technological interest in the recent years. In a recent experimental study, the Immm phase has been reported to be the thermodynamically stable phase of Li2C2under ambient conditions [1]. A previous density functional theory (DFT) based study of lithium carbide (Li2C2) predicted a pressure induced structural phase transition from orthorhombic (Immm) at 0 GPa → hexagonal (Bmmb) at 5GPa → cubic (I21 3)at 215GPa [4]. It was also reported that insulating ground state phase of Li2C2 with the band gap of 3.7eV turns metallic in the Bmmb phase, while I21 3 phase is again insulating with a band gap of 2.7eV. This interesting shift in the band gap and electronic properties of Li2C2 in three phases perused us to investigate the optical properties of the same in the three phases which are not yet reported. Furthermore being high band gap insulator Li2C2 is predicted to have useful optical properties for potential applications in optoelectronic devices.
The present work is intended to investigate in detail the electronic and optical properties of Li2C2 in Immm, Bmmb and I21 3 phases using full potential linearized augmented plane wave method plus local orbitals (FP-LAPW+lo) method
Computational Details
In order to investigate the structural, electronic and optical properties, the accurate FP-LAPW+lo method is used to solve Kohn–Sham equation within DFT [5] formulation as employed in the WIEN2k computer code [6]. The Engel-Voskogeneralized gradient approximation (EV GGA) [7] scheme is adopted to calculate the exchange-correlation energies. The charge density and potential were all expanded into two different basis sets. Inside the non-overlapping spheres surrounding the atomic sites (muffin-tin (MT) spheres), the potential was expanded into spherical harmonics with lmax = 10, while in the remaining (interstitial) regions the potential was expanded as plane waves. A plane wave cut off of Kmax = 7/RMT was used for the expansion of the wave function inside the interstitial regions; where RMT is the average radius of the MT spheres and Kmax is the maximum value of the wave vector K = k+G.
Results and Discussion
The band structure (BS) and density of states (DOS) of Li2C2 are calculated and shown in figures 1 and 2, respectively. It is clear from the DOS profile that the top of the valence band and the bottom of the conduction band for ‘Immm’ phase have a major contribution from hybridizations of ‘p’ and ‘d’ states of both ‘Li’ and ‘C’ atoms. Clearly, an indirect band gap of 4eV along ‘N’ to ‘Γ’ direction can be seen from the band structure profile shown in Fig. 2(a) for Immm phase.
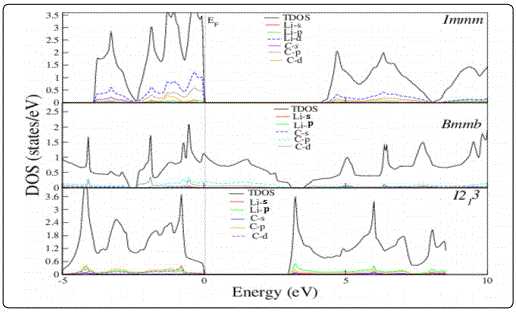
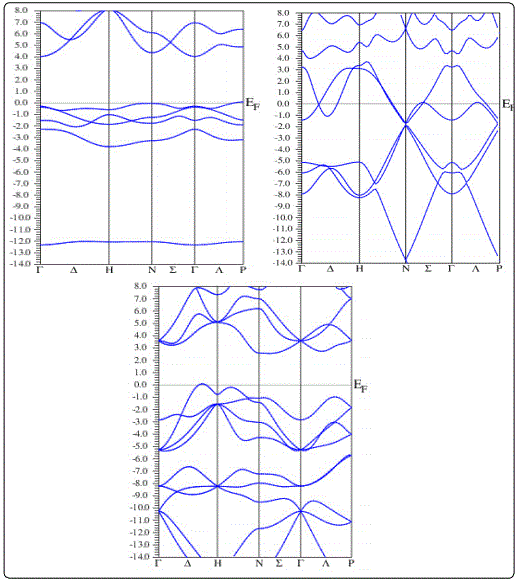
For Bmmb phase‘s’ and ‘p’orbitals of carbon forms three σ and one π band lying in the energy range from ‘-14eV to -2.5eV’. Furthermore, anti-bonding π band of carbon hybridizes with‘s’ and ‘p’ states of ‘Li’ and is dispersed from ‘-1eV’ (below the Fermi-level) to 2eV (above the Fermi-level)is responsible for the metallic character of Li2C2 in Immm phase. The metallic character is due to sp2 like hybrids in Immm phase. The cubic I21 3 againacquires an insulating nature with a band gap of 2.9eV due to diamond like sp3hybridization. Our calculated bandgap values for Li2C2 for Immm and I21 3 are in close agreement with earlier studies [4].
Optical properties are calculated in terms of dielectric function, reflectivity and optical conductivity. Dielectric functions of the ternary alloys are calculated using the following equations [8, 9].
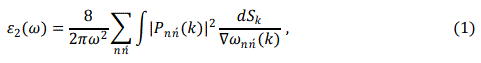
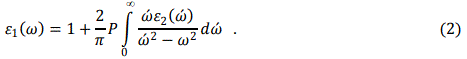
where Pnń(k) is the dipole matrix elements between initial and final states, Sk is an energy surface with constant value, wnń(k) is the energy difference between two states and p in Eq. 2, denotes the principal part of the integral.
The optical conductivity σ(ω) and normal incident reflectivity R(ω), are calculated using the following equations

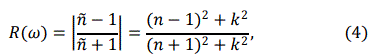
Where, Wev is transition probability per unit time.
Frequency dependent real and imaginary parts of the dielectric functions (ε1(ω)& ε2(ω)) of Li2C2 with electric field polarization along different crystallographic axis is shown in Fig.3. A considerable anisotropy in spectra of ε1(ω) & ε2(ω) for Immm and Bmmb is observed while I21 3 being cubic phase show no anisotropy. For Immm phase real part of the dielectric function for all electric field polarizations starts with a value between 2 to 4 at zero frequency and remains smooth up to energy below 5eV and then shows abrupt peaks at around 5eV which is linked with the fundamental band gap. Furthermore, it can be seen that the relative optical anisotropy (i.e. the difference between values of ε1(ω) for E⇀//x, E⇀// y and E⇀//z axis) increases with increase in energy. The dielectric function for I21 3 remains high in the energy range 4 to 9eV. On the basis of a high dielectric coefficient in the high UV range this material is highly suitable for use in opto-electronic devices.
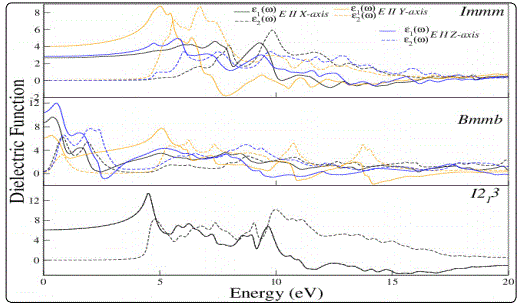
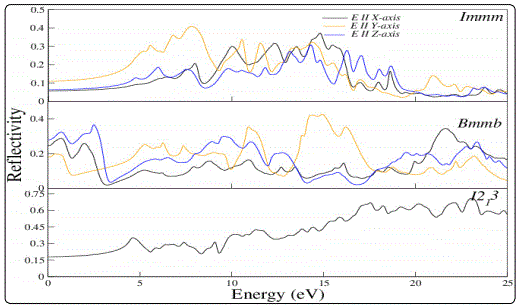
Reflectivity and optical conductivity of the compound in different phases is also studied. The reflectivity spectra are shown in Fig. 4. Reflectivity spectra of Li2C2 show different behaviour for Immm and I21 3 phases i.e. the reflectivity is maximum at about 5eV for Immm phase and keeps on decreasing with increase in energy on the other hand reflectivity for I21 3 phase is small at low energy, keep increasing and attains maximum value at 23eV energy. The optical conductivity spectra are shown in Fig. 5. Optical conductivity of Li2C2 for Immm and I21 3 phases remain high in the energy range ‘5 to 15 eV’. It may further be noted that the relative anisotropy for reflectivity and optical conductivity spectra increase with energy.
Conclusions
In summary, electronic and optical properties of Li2C2 are calculated using FPLAPW within DFT. Immm and I21 3 phases show insulating behaviour and Bmmb phase have metallic characteristics. The two phases Immm and I21 3 show high dielectric function and optical conductivity in the high UV range and are therefore suitable to be used in the fabrication of optoelectronic devices.
References
- Jiangtao H, Xiaoyan S, Wenwu X, Yuanyuan Z, Martin S, Markus R. Preparation and phase stability of nanocrystalline Li2C2 alloy. Material Letters. 2013; 94: 176–178. doi: 10.1016/j.matlet.2012.12.045
- Sang YL, Bong HB, Heun KK, et al. Reexamination of the structures and energies of Li2C2 and Li4C4. Chemical Physics Letters. 2005; 411(4-6): 484–491. doi: 10.1016/j.cplett.2005.05.123
- Martin D, Martin S, Artem K, Xiaoyan S, Rainer SF, Markus R. Thermodynamic stability of Li2C2 and LiC6. Journal of Alloys and Compounds. 2013; 575: 403-407. doi: 10.1016/j.jallcom.2013.06.001
- Kohn W, Sham LJ. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. A. 1965; 140: A1133. doi: 10.1103/PhysRev.140.A1133
- Engel E, Vosko SH. Exact exchange-only potentials and the virial relation as microscopic criteria for generalized gradient approximations. Phys. Rev. B. 1993; 47: 13164. doi: 10.1103/PhysRevB.47.13164
