

Research Article
The Multifaceted Mitochondria: Unexpected role of Mitochondria in Diseases
1Former, Associate Professor, S. D. N. B. Vaishnav College for Women, Chennai, India
2Former Director Grade Scientist, Centre for Cellular and Molecular Biology, Hyderabad, India
*Corresponding author: Purshottam Das Gupta, Centre for Cellular and Molecular Biology, Hyderabad, India, Tel: +918072891356, Email: pdg2000@hotmail.com
Received: January 14, 2022 Accepted: February 16, 2022 Published: February 22, 2022
Citation: Pushkala K, Gupta PD. The Multifaceted Mitochondria: Unexpected role of Mitochondria in Diseases. Madridge J Mol Biol. 2022; 3(1): 37-43. doi: 10.18689/mjmb-1000107
Copyright: © 2022 The Author(s). This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
A mitochondrion is a double-membrane-bound organelle found in most eukaryotic cells its primary function is to generate adenosine triphosphate, (ATP) which is utilized as a source of chemical energy by the organism. Lately in addition to generating ATP, they are involved several other functions according the requirement of the cell. In this review we described their role in proliferation, differentiation, information transfer, apoptosis and role in therapeutics. Since they have their own DNA they also take part in protein synthesis; part of the key enzyme-, Succinic- dehydrogenase-is synthesized under mitochondrial genome. Mitochondrial myopathies are a group of neuromuscular disease caused by damage of the mitochondria with some examples including Kearns-Sayre Syndrome (KSS), Leighʼs syndrome, Mitochondrial Depletion Syndrome (MDS), Mitochondrial ephalomyopathy, Lactic Acidosis and Stroke-like episodes (MELAS). Scientist took advantage of the fact that in all human population mitochondria comes from mother only and therefore they could trace of human origin and then migration. Lately, Mitochondrial replacement therapy and Mitochondrial diffusion therapies were developes and treated rere diseases which had no treatment prior to this development.
Keywords: Porin proteins; Adenosine triphosphate (ATP); Information transfer; Apoptosis, Mitochondrial myopathies; Mitochondrial replacement therapy and Diffusion therapies.
Introduction
Mitochondria semi autonomous organelle lodged in almost all eukaryotic cells outside of the nucleus; present in few numbers to several thousand per cell. Mitochondria are spherical, double-membrane-bound organelle with specific function for their membranes. Mitochondrial outer membrane permeability is conferred by a family of porin proteins. Mitochondrial porins conduct small molecules and constitute one component of the permeability transition pore that opens in response to apoptotic signals [1]. The porins in the outer membrane allow the passage of molecules smaller than 5 kilodaltons besides the large multiprotein translocase complex that recognizes mitochondrial signal sequences on larger proteins and permits their passage. The matrix, located inside the inner membrane has mitochondrial DNA (mtDNA) [2].
The tissue with increased energy demand has more mitochondria (for e.g. heart muscle cells). Also the inner membrane of the organelle has many folds in the form of finger like processes the cristae, in places to increase the surface area for energy production [3]. Endo symbiosis theory claims mitochondria to have evolved from purple bacteria approximately 1.5 billion years ago [4]. Chloroplasts in plant cells are the only cell organelles with DNA though generally this genetic molecule is found in the nucleus having striking similarities to bacteria cells. Recently it has been proved that mitochondria are also involved in many other functions in addition to energy production to meet the cellular demand. This “powerhouse” of the cell manufactures ATP and other metabolites, is also a “sentinel” organelle capable of both detecting cellular insults and orchestrating inflammatory responses [5].
Inheritance of mitochondria to the progeny is through mother and, thus, can be traced back to a single female ancestor. The maternal ancestor of all living humans confirms that “Mitochondrial Eve” lived about 200,000 years ago. The DNA in the mitochondria is solely from the ooplasm and not from the sperm and so mtDNA is matrilineal [6-9]. It is interesting to note that pattern of human migration can be traced due to mtDNA mutation [10]. The mtDNA is neither enveloped nor packaged into chromatin. Both mitochondria and chloroplast use their DNA to produce many proteins and enzymes required for their function [11].
Therefore, the aim of this review is to discuss current knowledge of how mitochondrial structure can be modulated and coordinated across different spatial scales to support cells facing diverse functional demands. Highlighting how mitochondrial structure is tightly regulated within a cell, we focus on the functional tradeoffs associated with structural alterations within a mitochondrion, at the single organelle and mitochondrial network levels, and between mitochondria and other organelles which contribute to cellular functional specificity.
Functions in additional to energy production
Proliferation, differentiation, information transfer, apoptosis and role in therapeutics are additional involvement by the mighty mitochondria. In order to achieve these functions, the mitochondria need to move to the corresponding location inter and intracellular. This transcellular transfer of mitochondria is dynamically involved in the cellular and tissue response to CNS injury and play beneficial roles in recovery [12]. Mitochondria primarily move by the action of molecular motors along cytoskeletal elements. Like other organelles, mitochondria associate with specific motor isoforms through organellespecific adaptors, and their movement is sensitive to disruption of these motors and adaptor proteins [13]. Therefore, mitochondrial movement has a crucial role in normal physiologic activity, and any disorder in the movement will cause irreparable damage to the organism.
Human life span could be associated with the life length of the mother but not the father, suggesting an influence of the maternal inherited mitochondrial genome [14]. Therefore, mitochondria-associated disease mutations are also always inherited maternally.
Human circular mtDNA was the first significant part of the human genome sequenced and was shown that it contains 16,569 base pairs 37 mitochondrial (mt) genes including 13 coding for essential components of the mitochondrial electron transport chain and of the ATP synthase complex, 22 for mitochondrial transfer RNAs and 2 for ribosomal RNAs. mtDNA codes for the genetic information required by mitochondria whereas nuclear DNA is encoded for the genetic information required by the entire cell [15,16]. One mitochondrion contains dozens of copies of its mitochondrial genome in contrast to one copy of its nuclear genome [2]. It is found that 3% of the mitochondrial genome is non coding compared to 93% of the nuclear genome. Some mitochondrial coding sequences (triplet codons) do not follow the universal codon usage rules when they are translated into proteins. The same nucleotide can sometimes function as both the last base of one gene and the first base of the next gene thereby bases exhibiting functional overlap between two genes in mitochondria. Mitochondrial genes on both DNA strands are transcribed in a polycistronic manner [17]. Large mitochondrial mRNAs contain the instructions to build many different proteins, which are encoded one after the next along the mRNA differing from nuclear genes which are usually transcribed one at a time from their own mRNA. The dysfunction of any type is observed to be the causal factor for the development of many medical problems. The mtDNA is more susceptible to damage than the rest of the genome due to the free radicals produced during ATP synthesis [2].
The mitochondria contain over 100 components of the respiratory chain, 13 of which are coded by mtDNA, the remaining being encoded by nuclear DNA physically interlocked like pieces of a jigsaw. The data of the study also provided a clue that genetic variants in mtDNA passed to offspring could increase the risk of developing different conditions, as well as influence characteristics such as height and lifespan [18]. Genetic longevity studies provides enough evidence for life length could be associated with the life length of the mother but not the father, suggesting an influence of the maternal inherited mitochondrial genome entire life-course of individuals in humans [14]. Therefore, mitochondria-associated disease mutations are also always inherited maternally. mtDNA is a proper tool for the determination of the origin of populations due to its high evolutionary importance. Ancient mitochondrial DNA retrieved from museum specimens, archaeological finds and fossil remains can provide direct evidence for population origins and migration processes [19].
Protein synthesis
Mitochondria are no longer known simply as the powerhouse of the cell as they have now been shown to play a key part in several cellular processes including metabolite biosynthesis beyond their classical role in energy metabolism. While it is becoming increasingly well established that mitochondrial protein composition can vary largely among different cell types [20-24] as well as how mitochondrial structural alterations contribute to the wide range of mitochondrial functional capacities observed in different cells is less well understood. In the rabbit heart three days before birth, the matrix makes up 48% of mitochondrial volume while the inner boundary membrane + cristae take up 39%. However, two days after birth, the transition to more aerobic metabolism results in a redistribution of mitochondrial volume where the inner membrane and cristae occupy more volume than the matrix (47 vs. 42%, respectively) [25].
In support of this increased cellular synthesis demand, the mitochondria in the liver have a relatively greater capacity for metabolite biosynthesis and lower energy conversion capabilities compared to striated muscle mitochondria [21,26]. Using histochemical techniques for the first time in Bufo by Ward [27] presented evidence for the origin of protein crystals within a single crista in some oocyte mitochondria. Later Gupta [28] also showed protein yolk synthesis in invertebrate oocytes. In Neurospora crassa mitochondria takes part in the synthesis of one subunit of the succinic dehydrogenate enzyme [29].
Mitochondrial DNA mutation (mtDNA) and its significance
Mutation rate of mtDNA is 100 fold than the nuclear genome resulting in the presence of heterogeneous population of mtDNA within the same cell, and even within the same mitochondrion. During cell division daughter cells receive similar, but not identical, copies of their mtDNA. A nuclear gene, called DNA polymerase gamma (POLG), encodes the DNA polymerase responsible for replicating the mitochondrial genome. This enzyme with two domains: a catalytic domain that exhibits polymerase activity, and an exonuclease domain that is involved in the recognition and removal of DNA base-pair mismatches that occur during DNA replication. The higher mutation rate is correlated with a nucleotide imbalance that leads to decreased (POLG), fidelity [30].
Mutations of any type in mtDNA lead to a subtle differences in our ability to produce energy. These differences in the power house of the cell affect various complex biological pathways inside our systems, where the signals that allow our cells to operate in a coordinated fashion. The functions of this organelle are critical to cell survival in a variety of ways. For instance, they help in storage of calcium ions as well as calcium transport and signalling [31,32].
The role of mitochondria in the defence mechanism is significant. When a macrophage engulfs bacteria, it triggers a stress pathway in the endoplasmic reticulum which in turn stimulates mitochondria to produce reactive oxygen species (ROS) which are packaged into vesicles and shuttled to the phagosome. The damaging molecules are thought to aid in killing the pathogen. Finally, the left over particles of the bacterium are degraded once the phagosome fuses with a lysosome [33]. Due to the presence of DNA as shown by staining technique, protein synthesis is also possible within the mitochondria as discuss earlier [34,35]. However, for the first time we have shown that one component of succinic dehydrogenase, the marker enzyme of the mitochondrion, is synthesized under nuclear genome in the cytoplasm from where it is translocated through endoplasmic reticulum into the inner membrane of the mitochondria and the synthesis second component of the enzyme is by mitochondrial genome. Thus both the nuclear genome and mtDNA involve together during the synthesis of the enzyme [36].
Mitochondria affect health and development
Maintaining calcium in right concentration is indispensible for blood clotting [37], muscle contraction [38] and other important tasks. Apart from making iron compounds carry oxygen to tissues they are in the initial production site for steroid hormones including cortisol, estrogen, progesterone and testosterone [39].
Mitochondria play a central role in initiation of the intrinsic pathway of apoptosis by releasing mitochondrial proteins, which normally reside in the inter membrane space into the cytosol [40-42]. It has also been demonstrated that aging human colonic cells displaying respiratory chain deficiency have a significant higher apoptotic frequency compared to normal human colonic cells indicating that respiratory deficiency induces apoptosis.
Mitochondrial myopathies are a group of neuromuscular disease caused by damage of the mitochondria with some examples including Kearns-Sayre Syndrome, Leighʼs syndrome, Mitochondrial Depletion Syndrome, Mitochondrial Encephalomyopathy, Lactic Acidosis and Stroke-like episodes [43-44] in humans.
mtDNA is more susceptible to damages in comparison to nuclear DNA. Importantly, mtDNA molecules are not protected by histones and supported with only rudimentary DNA repair mechanism. Besides they are localized in close proximity to the electron transport chain, which continuously generates Reactive Oxygen Species (ROS). As a result mutation rate of mtDNA has been reported to be up to 15-fold higher than nuclear DNA in response to DNA damaging agents. The mechanisms described encompass altered production of mitochondrial ROS, altered regulation of the nuclear epigenome, affected initiation of apoptosis, and a limiting effect on the production of ribonucleotides and deoxyribonucleotides [45].
Inherited changes in mtDNA can also cause problems with growth, development, and function of the bodyʼs systems. These mutations disrupt the mitochondriaʼs ability to generate energy efficiently for the cell. Conditions caused by mutations in mtDNA often involve multiple organ systems. The effects of these conditions are most pronounced in organs and tissues that require a lot of energy, such as the heart, brain, and muscles [44].
Recently it was shown that mitochondrial dysfunction in placental trophoblast cells prove to be a causal factor for gestational diabetes mellitus. This is in part complicated by the different mitochondrial subpopulations present in the two major trophoblast cell lineages of the placenta. A study examined key aspects of mitochondrial function in placentas from healthy pregnancies and those complicated gestational diabetes mellitus (GDM) in both whole tissue and isolated mitochondria. Mitochondrial content, citrate synthase activity, ROS production and gene expression regulating metabolic, hormonal and antioxidant control was examined in placental tissue, before examining functional differences between mitochondrial isolates from cytotrophoblast and syncytiotrophoblast. The observations indicated the mitochondrial dysfunction across multiple pathways when assessing whole placental tissue from GDM pregnancies compared with healthy controls [46].
Somatic mutations in mtDNA have been associated with some forms of cancer and an increased risk of certain age-related disorders such as heart disease, Alzheimer disease, and Parkinson disease. Accumulations of somatic mutations in a personʼs lifetime may play a role in the normal process of aging. It is conceivable that mitochondrial deficiency could lead to mutagenesis in the nuclear genome also [47]. It is also possible that mutation may occur in the mitochondrial genes of sperm but not in the blood cells [48]. Mitochondria play a cyclic role in the generation of steroid hormone estrogen) that in turn, modulate mitochondrial activities. The impairment of this organelle was observed to be one of the central features of aging in women via neuroprotective, neurotrophic and antioxidant modes of action. The hypoestrogenic state in the peri- as well as in the prolonged postmenopause might increase the vulnerability of elderly women to brain degeneration and age-related pathologies. A number of recent studies link mitochondrial function to signalling pathways that regulate brain plasticity, life span and to the aging process. In this context, estrogen/ Brain derived neurotrophic factor (BDNF) or estrogen/ Sirtuin 3 (SIRT3) actions and interactions represent complex and fundamental mechanisms of neuronal plasticity, a process highly depending on energy supply via mitochondrial activity [49].
Somatic mutations of the mtDNA though not inherited by the next generation, gets accumulated over time due to their inability to repair it when it is damaged. Interestingly, high levels of mtDNA mutations have been found in many tumours and cancer cells [45,47,50,51].
In human, several neurodegenerative disorders are correlated with mitochondrial dysfunction and oxidative damage leading to major neuronal loss. Free radicals, typically generated from mitochondrial respiration, cause oxidative damage of nucleic acids, lipids, carbohydrates and proteins. A common features of toxicity observed that are related to oxidative damage responsible for Huntingtonʼs disease, Friedreich ataxia and Xeroderma pigmentosum, provide insight into shared mechanisms of neuronal death [52]. In the case of Huntingtonʼs disease oxidative DNA damage can cause pathway for repairing oxidative base lesions to expand trinucleotide repeats. Accumulation of oxidative mtDNA damage during aging is associated with Alzheimerʼs disease, Parkinsonʼs disease and amyotrophic lateral sclerosis. Many human diseases such as muscle disuse/inactivity, diabetes, cancer, renal, and cardiac failure and in aging-sarcopenia are associated with catabolic conditions such as loss of muscle mass and force leading to alternation in the mitochondrial content, morphology and functions. Mitochondria were suspected to affect negatively by amyloid β peptide (Aβ), an important component in Alzheimerʼs disease pathogenesis, a causal factor for mitochondrial dysfunction and oxidative stress. The progressive accumulation of mitochondrial Aβ is associated with aberrant mitochondrial functions leading to neuronal damage and cognitive decline Aβ is transported into mitochondria via the translocase of the outer membrane TOM import machinery localized to mitochondrial cristae [53-57].
Mitochondrial dysfunction has been observed in several of the diseases that were associated with mitochondrial single nucleotide variants such as multiple sclerosis, type II diabetes and abdominal aortic aneurysms and many syndromes [58].
The changes of mitochondrial network influence the production of ROS that play an important role in muscle function. Moreover, dysfunctional mitochondria trigger catabolic signalling pathways which feed-forward to the nucleus to promote the activation of muscle atrophy [59].
Loss of functional mitochondrial complex I (MCI) in the dopaminergic neurons of the substantia nigra is a hallmark of Parkinsonʼs disease. In mice model, MCI dysfunction alone is sufficient to cause progressive parkinsonism similar to human in which the loss of MCI in the dopaminergic neurons of the substantia nigra [60]. Multiple lines of evidence, including clinical, genetic, ultrastructural, and biochemical studies, support the involvement of mitochondria in the patho-physiology of psychiatric illness [61].
Mitochondrial replacement therapy
The first case of suspected mitochondrial disease occurred during 1962, in a woman with extremely fast and efficient metabolism. Her muscle tissue had large size as well as more number of mitochondria. Subsequently, mitochondrial dysfunction associated with mtDNA mutations leading to diseases, including seizure, ataxia, cortical blindness, dystonia, exercise intolerance, ophthalmoplegia, optic atrophy, cataracts, diabetes mellitus, short stature, cardiomyopathy, sensorineural hearing loss and kidney failure [9,43,62-64]. The technical advancement paved way for the replacement of defective mitochondria of the mother with a donor, thereby protecting her child from having a potentially life-threatening mitochondrial disease.
MRT or Mitochondrial Gene Therapy (MGT) is a medical technique where defective mitochondria carried by a woman are replaced with the healthy mitochondria of a donor. MRT has been associated with a number of terms, some of which conveyed positive implications like “Mitochondrial gene therapy”, “Mitochondrial donation”, “Life-saving Treatment”, “Narratives of Hope” while some others made negative impacts like “Three parent baby”, “Three-person baby”, “Three persons DNA”, “Slippery Slope”, “Designer babies”. The truth is that it is the nuclear DNA around which the whole concept of childʼs genetic identity and personality revolves since, nuclear DNA is the one to make a profound impact on the latter, not the mtDNA [9]. Mitochondrial Replace Therapy also referred to as “Mitochondrial Donation technique” [65] associated with the category of techniques in which the embryo possessing the nuclear DNA of the parents is subjected to in vitro fertilization (IVF) procedure to have mtDNA of the donor female [66]. MRT include different techniques like spindles transfer, pronuclear transfer or polar body transfer [67-70].
In Pronuclear transfer technique: Two zygotes are raised in vitro. One belongs to the biological parents with pronuclei and defective mitochondria and the second one having pronuclei and healthy mitochondria [71]. The pronuclei of biological parents are taken out and transplanted into the donorʼs zygote (with rejected pronuclei) with healthy mitochondria by using electric pulses or inactivated hemagglutinating virus of Japan [72] The reformed zygote is transferred to the motherʼs womb [73]. The worldʼs first, three-parent baby (boy) was born, showing no signs of genetic disorder on 6th April 2015 [74].
Maternal spindle transfer (MST) technique
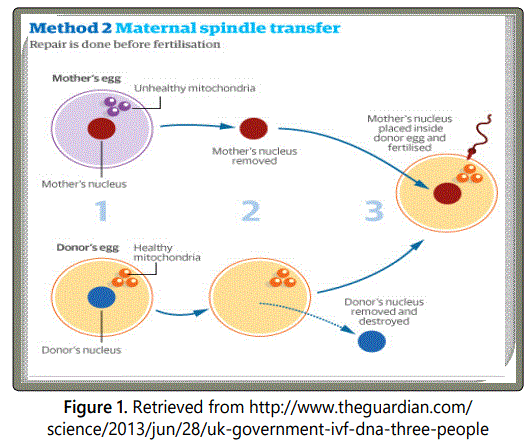
Maternal spindle transfer technique has been adopted and successfully executed by Dr. John Zhang and his team [75]. The technique executed before fertilization is a form of selective reproduction similar to prenatal diagnosis and pre-implantation genetic diagnosis [76]. The maternal spindle complex at the metaphase stage is extracted from the defective egg of the mother, which is then transplanted into the perivitelline space of the enucleated donorʼs egg with healthy mitochondria (Figure 1) [77]. The reformed embryo is transplanted into the motherʼs womb. This approach is preferable because maternal spindle contains little cytoplasm which eventually reduces the chances of mtDNA carryover and mutations [78].
Polar body genome transfer (PBT) It is considered as the most significant approach because of the presence of the scarce mitochondria with little cytoplasm which minimizes the possibilities of mtDNA carryover. All the nuclear content enclosed within polar bodies increases its potency to confer the re-established oocytes and zygotes by the above two methods. The idea of the usage of the polar bodies was first put forth by Wakayama and Yanagimachi but adopted by Wang and his colleagues [79] to perform the technique in mice where, the transfer of first and the second polar body led to the normal progression of the progenies [80].
Prons and Cons of the therapy
It would be unfair to females with mitochondrial disease to limit their conceptive range [81]. Apart from biological relations, there is another relation that exists between mother and child, an emotional connection.
There are minimal chances of risks correlated with the mixing of prospective motherʼs mtDNA and donor mtDNA [82]. The proofs about discrepancies are not reported yet. It is feasible to match the haplotypes of the two [83]. There are little possibilities of mtDNA alterations and is unlikely to be troublesome [84].
Egg donation can lead to ovarian hyper stimulation syndrome and its genetic kinship cannot be achieved as the whole gamete (nuclear and mtDNA is shared by a third person [71,85]. Prenatal diagnosis is unsuccessful in heteroplastic populations. Pre-implantation Genetic Diagnosis is appropriate for women with low levels of deficient mtDNA [86]. The drawbacks of these alternatives illustrate the importance of MRT for couples with mitochondrial diseases.
Mitochondrial infusion
Like cell infusion technique, more recently 2018, Dr. Sitaram Emani and his colleagues have restored dying organs to Life by infusing mitochondria into a blood vessel feeding the heart, instead of directly into the damaged muscle. Somehow the organelles will gravitate almost magically to the injured cells that need them and take up residence.
References
- Weeber EJ, et al. The role of mitochondrial porins and the permeability transition pore in learning and synaptic plasticity. J Biol Chem. 2002; 277(27): 18891-7. doi: 10.1074/jbc.M201649200
- Mighty Mitochondria. 2014; Biomedical Beat Blog.
- Druzhyna NM, Wilson GL, LeDoux SP. Mitochondrial DNA repair in aging and disease. Mech Ageing Dev. 2008; 129(7-8): 383-90. doi: 10.1016/j.mad.2008.03.002
- Li X, Fang P, Mai J, Choi ET, Wang H, Yang XF. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J Hematol Oncol. 2013; 6: 19. doi: 10.1186/1756-8722-6-19
- Mothers can influence offspringʼs height, lifespan and disease risk through mitochondria. 2021; University of Cambridge.
- Cyran KA, Kimmel M. Alternatives to the Wright–Fisher model: The robustness of mitochondrial Eve dating. Theor Popul Biol. 2010; 78(3): 165-72. doi: 10.1016/j.tpb.2010.06.001
- Kali. Wikipedia.
- Gupta PD. Mitochondrial Eve: Its Genetic Implications. J Genet Syndr Gene Ther. 2020; 11: 334.
- Witas HW, Zawicki P. Mitochondrial DNA and human evolution: a review. Anthropol Rev. 2004; 67: 97-110.
- The Genetic Systems of Mitochondria and Plastids. 2002; 4th edition. Molecular Biology of the Cell.
- Shanmughapriya S, Langford D, Natarajaseenivasan K. Inter and Intracellular mitochondrial trafficking in health and disease. Ageing Res Rev. 2020; 62: 101128. doi: 10.1016/j.arr.2020.101128
- Kruppa AJ, Buss F. Motor proteins at the mitochondria-cytoskeleton interface. J Cell Sci. 2021; 134: jcs226084. doi: 10.1242/jcs.226084
- Niels van den Berg, Mar Rodríguez-Girondo, Anton J M de Craen, et al. Longevity around the Turn of the 20th Century: Life-Long Sustained Survival Advantage for Parents of Todayʼs Nonagenarians. J Gerontol A Biol Sci Med Sci. 2018; 73(10): 1295-1302. doi: 10.1093/gerona/gly049
- Fernández-Silva P, Enriquez JA, Montoya J. Replication and Transcription of Mammalian Mitochondrial DNA. Exp Physiol. 2003; 88(1): 41-56. doi: 10.1113/eph8802514
- Fazzini F, Schopf B, Blatzer M, et al. Plasmid-normalized quantification of relative mitochondrial DNA copy number. Sci Rep. 2018; 8: 15347. doi: 10.1038/s41598-018-33684-5
- Mercer TR, Neph S, Dinger ME, et al. The human mitochondrial transcriptome. Cell. 2011; 19: 146(4): 645-658. doi: 10.1016/j.cell.2011.06.051
- Yonova-Doing E, Calabrese C, Gomez-Duran A, et al. An atlas of mitochondrial DNA genotype–phenotype associations in the UK Biobank. Nat Genet. 2021; 53(7): 982-993. doi: 10.1038/s41588-021-00868-1
- Nesheva DV. Aspects of ancient mitochondrial DNA analysis in different populations for understanding human evolution. Balkan J Med Genet. 2014. 17(1): 5-14. doi: 10.2478/bjmg-2014-0019
- Mootha VK, Bunkenborg J, Olsen JV, et al. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell. 2003; 115(5): 629-40. doi: 10.1016/s0092-8674(03)00926-7
- Johnson RB, Onwuegbuzie AJ, Turner LA. Toward a definition of mixed methods research. Journal of Mixed Methods Research. 2007; 1(2): 112-133. doi: 10.1177/1558689806298224
- Pagliarini DJ, Calvo SE, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008; 134(1): 112-123. doi: 10.1016/j.cell.2008.06.016
- Calvo SE, Clauser KR, Mootha VK. MitoCarta2. 0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016; 44: D1251-D1257. doi: 10.1093/nar/gkv1003
- Benador IY, Veliova M, et al. Mitochondria bound to lipid droplets have unique bioenergetics, composition, and dynamics that support lipid droplet expansion. Cell metabolism. 2018; 27(4): 869-885.e6. doi: 10.1016/j.cmet.2018.03.003
- Smith HE, Page E. Ultrastructural changes in rabbit heart mitochondria during the perinatal period: neonatal transition to aerobic metabolism. Dev Biol. 1977; 57(1): 109-117. doi: 10.1016/0012-1606(77)90358-x
- Phillips D, Covian R, et al. Regulation of oxidative phosphorylation complex activity: effects oftissue-specific metabolic stress within an allometric series and acute changes in workload. Am J Physiol Regul Integr Comp Physiol. 2012; 302(9): R1034-R1048.
- Ward RT. The origin of protein and fatty yolk in Rana pipiens. II.Electron microscopical and cytochemical observations of young and mature oocytes. J Cell Biol. 1962; 14(2): 309-41. doi: 10.1083/jcb.14.2.309
- Koke JR, Gupta PD, Malhotra SK. Succinic dehydrogenase activity in “Mesosomes” of Neurospora crassa. Biochemical and biophysical research communications. 1971; 42: 576-582. doi: 10.1016/0006-291x(71)90410-4
- Song S, et al. DNA precursor asymmetries in mammalian tissue mitochondria and possible contribution to mutagenesis through reduced replication fidelity. Proc Natl Acad Sci. 2005; 102(14): 4990-4995. doi: 10.1073/pnas.0500253102
- Simona Boncompagni, et al. Mitochondria are linked to calcium stores in striated muscle by developmentally regulated tethering structures. Mol Bio Cell. 2009; 20(3): 1058-1067. doi: 10.1091/mbc.e08-07-0783
- Bravo-Sagua R, et al. Calcium transport and signalling in mitochondria. Compr Physiol. 2017; 7(2): 623-634. doi: 10.1002/cphy.c160013
- Abuaita BH, et al. Mitochondria-derived vesicles deliver antimicrobial reactive oxygen species to control phagosome-localized Staphylococcus aureus. Cell Host Microbe. 2018; 24(5): 625-636. doi: 10.1016/j.chom.2018.10.005
- Gupta PD, Koke JR, Malhotra SK. Remarks on membranes of Neurospora crassa after embedding in glycol methacrylate. Cytobios. 1971; 3(10/11): 117-128.
- Barile CJ, Herrmann PC, et al. Inhibiting platelet-stimulated blood coagulation by inhibition of mitochondrial respiration. Proc Natl Acad Sci USA. 2012; 109(7): 2539-2543. doi: 10.1073/pnas.1120645109
- Zhou J, Dhakal K, Yi J. Mitochondrial Ca(2+) uptake in skeletal muscle health and disease. Sci China Life Sci. 2016; 59: 770-776. doi: 10.1007/s11427-016-5089-3
- Miller WL, Auchus RJ. The Molecular Biology, Biochemistry, and Physiology of Human Steroidogenesis and Its Disorders. Endocr Rev. 2011; 32(1): 81-151. doi: 10.1210/er.2010-0013
- Luciano L, Gupta PD, et al. Modulation of apoptosis by starvation: morphological and biochemical study of rat intestinal mucosa. Cell Death Differ. 1995; 2(4): 259-66.
- Tsou KC, Mela L, Gupta PD, Lynm D. Mitochondrial ultrastructure study with the new cytochrome c binding agent 4,4′-diamino-2,2′-bipyridyl ferrous chelate. J Ultrastructure Research. 1976; 54(2): 235-242. doi: 10.1016/S0022-5320(76)80153-0
- Fisher JJ, Vanderpeet CL, et al. Mitochondrial dysfunction in placental trophoblast cells experiencing gestational diabetes mellitus. J Physiol. 2021; 599: 1291-1305. doi: 10.1113/JP280593
- Thangaraj K, et al. Sperm mitochondrial mutations as a cause of low sperm motility. J Androl. 2003; 24(3): 388-392. doi: 10.1002/j.1939-4640.2003.tb02687.x
- Lejri1 I, Grimm1 A, Eckert A. Mitochondria, Estrogen and Female Brain Aging. Front Aging Neurosci. 2018; 27(10): 124. doi: 10.3389/fnagi.2018.00124
- Penta JS, Johnson FM, et al. Mitochondrial DNA in human malignancy. Mutat Res. 2001; 488: 119-133.
- Chatterjee A, Mambo E, Sidransky D. Mitochondrial DNA mutations in human cancer. Oncogene. 2006; 25: 4663-4674. doi: 10.1038/sj.onc.1209604
- Trushina E, Mc Murra CT. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience. 2007; 145: 1233-1248.
- Cleaver JE, Lam ET, Revet I. Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity. Nat Rev Genet. 2009; 10(11): 756-768. doi: 10.1038/nrg2663
- Jonson I, Ougland R, Larsen E. DNA Repair Mechanisms in Huntingtonʼs Disease. Molecular Neurobiology. 2013; 47: 1093-1102. doi: 10.1007/s12035-013-8409-7
- Jeppesen DK, Vilhelm A, Bohr VA, Stevnsner T. DNA Repair Deficiency in Neurodegeneration. Prog Neurobiol. 2011; 94(2): 166-200. doi: 10.1016/j.pneurobio.2011.04.013
- Guo C, Sun L, Chen X, Zhang D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen Res. 2013; 8: 2003-2014. doi: 10.3969/j.issn.1673-5374.2013.21.009
- Picone P, Nuzzo D, Caruana L, et al. Mitochondrial Dysfunction: Different Routes to Alzheimerʼs Disease Therapy. Oxid Med Cell Longev. 2014; 2014: 780179. doi: 10.1155/2014/780179
- Joshua JF, Chelsea LV, et al. Mitochondrial dysfunction in placental trophoblast cells experiencing gestational diabetes mellitus. J Physiol. 2021; 599(4): 1291-1305. doi: 10.1113/JP280593
- Romanello V, et al. Mitochondrial Quality Control and Muscle Mass Maintenance. Front Physiol. 2015; 6: 422. doi: 10.3389/fphys.2015.00422
- González-Rodríguez P, et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature. 2021; 599(7886): 650-656. doi: 10.1038/s41586-021-04059-0
- Anglin RES, Mazurek MF, Tarnopolsky MA, Rosebush PI. The Mitochondrial Genome and Psychiatric Illness. Am J Med Genet B Neuropsychiatr Genet. 2012; 159B(7): 749-59. doi: 10.1002/ajmg.b.32086
- Schulmann A, Ryu E, et al. Novel Complex Interactions between Mitochondrial and Nuclear DNA in Schizophrenia and Bipolar Disorder. Mol Neuropsychiatry. 2019; 5: 13-27. doi: 10.1159/000495658
- Park A, et al. Mitochondrial Transplantation as a Novel Therapeutic Strategy for Mitochondrial Diseases. Int J Mol Sci. 2021; 22: 4793. doi: 10.3390/ijms22094793
- Lu J, Lokendra KS. Implications of mitochondrial DNA mutations and mitochondrial dysfunction in tumorigenesis. Cell Research. 2009; 19(7): 802-815. doi: 10.1038/cr.2009.69
- Wolf DP, Mitalipov N, Mitalipov S. Mitochondrial replacement therapy in reproductive medicine. Trends Mol Med. 2015; 21(2): 68-76. doi: 10.1016/j.molmed.2014.12.001
- Sharma H, et al. Development of mitochondrial replacement therapy: A review. Heliyon. 2020; 6: e04643.
- Kaur S, Nagpal M. Recent advancement in human reproduction threeparent babies: a technique to neutralize mitochondrial disease load- A boon or a bane for society?. Curry Trends Diagn Treat. 2017; 1(2): 100-103. doi: 10.5005/jp-journals-10055-0024
- Amato P, et al. Three-parent in vitro fertilization: gene replacement for the prevention of inherited mitochondrial diseases. Fertil Steril. 2014; 101(1): 31-35. doi: 10.1016/j.fertnstert.2013.11.030
- Herbert M, Turnbull D. Progress in mitochondrial replacement therapies. Nat Rev Mol Cell Biol. 2018; 19(2): 71-72. doi: 10.1038/nrm.2018.3
- Wringley A, et al. Mitochondrial replacement: ethics and identity. Bioethics. 2015; 29(9): 631-638.
- Labarta E, et al. Mitochondria as a tool for oocyte rejuvenation. Fertil Steril. 2019; 111(2): 219-226. doi: 10.1016/j.fertnstert.2018
- Wang K, et al. CARL lncRNA inhibits anoxia-induced mitochondrial fission and apoptosis in cardiomyocytes by impairing miR-539-dependent PHB2 down regulation. Nat Commun. 2014; 5: 1-13.
- Chiang JL, Shukla P, Pagidas K, et al. Mitochondria in ovarian aging and reproductive longevity. Ageing Res Rev. 2020; 63: 101168. doi: 10.1016/j.arr.2020.101168
- Chinnery PF, Carven L, et al. The challenges of mitochondrial replacement. PLoS Genet. 2014; 10(4): e1004315. doi: 10.1371/journal.pgen.1004315
- Dimond R. Ethics of mitochondrial gene replacement therapy. Crossroads of Genetic and Reproductive echnologies. 2018; 31-53. doi: 10.1016/B978-0-12-813764-2.00002-7
- Dimond R, Stephens N. Three persons, three genetic contributors, three parents: mitochondrial donation, genetic parenting and the immutable grammar of the ‘three xx. Health. 2017; 22(3): 240-258. doi: 10.1177/1363459316689380
- Pickett SJ, et al. Mitochondrial Donation- Which women could benefit?. N Engl J Med. 2019; 380: 1971-1972. doi: 10.1056/NEJMc1808565
