

Research Article
Using of Cytoscape Build Large Scale interface Bionetwork and Analysis Topological Distinctiveness of PPIs of Mycobacterium Tuberculosis H37Rv
1Department of Computational Biology and Bioinformatics, Jacob Institute of Biotechnology and Bioengineering, Sam Higginbottom University of Agriculture, Technology and Sciences, Allahabad, Uttar Pradesh, India
2RASS Biosolutions Pvt. Ltd., Civil Lines, Kanpur, Uttar Pradesh, India
*Corresponding author: Ved Kumar Mishra, Department of Computational Biology and Bioinformatics, Sam Higginbottom University of Agriculture Technology and Sciences Allahabad, Uttar Pradesh, India, E-mail: ved.m45@gmail.com
Received: January 2, 2019 Accepted: February 25, 2019 Published: March 8, 2019
Citation: Mishra VK, Jain PA, Khanna S. Using of Cytoscape Build Large Scale interface Bionetwork and Analysis Topological Distinctiveness of PPIs of Mycobacterium Tuberculosis H37Rv. Madridge J Mol Biol. 2019; 1(1): 4-13. doi: 10.18689/mjmb-1000102
Copyright: © 2019 The Author(s). This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Bioinformatics open software tool cytoscape is worn for the visualizing and integrating gene expression of molecular interaction networks. Protein–protein interactions (PPIs) form the foundation for an enormous mainstream of cellular events, together with signal transduction and transcriptional regulation. It is currently implicit so as to the swot up of interactions and communications among cellular macromolecules is fundamental to the indulgent of biological systems. Interactions among proteins have been premeditated all the way during a number of elevated-throughput experiments. It has furthermore been predicted from side to side an assortment of computational process so as to leverage the immense quantity of sequence data which generate in the previous decade. In the current research we used an unfasten basis software cytoscape for integrating the biomolecularinteraction networks with high throughput expression data. Molecular states an amalgamated conceptual framework on behalf of the forecast of efficient linkages among proteins. In this research work there is epigrammatic explanation of the databases and PPIs tools. In this work explanation of a preface to network theory and important network topologies parameters universally used in analyzing networks, over and above process to recognize significant network apparatus, based on perturbations.
Keywords: CNS; Cytoscape; ATC; NCBI; Pubchem.
Introduction
An oldest identified human disease Tuberculosis (TB) is still one of the foremost responsible of mortality, in view of the fact that near about two million citizens depart this life to death every year commencing this trouble. TB has countless manifestations, distressing bone with the central nervous system (CNS), and various erstwhile organ systems, excluding it is first and foremost a pulmonary disease i.e., initiated via the authentication of Mycobacterium tuberculosis. The evolution of the infection can have more than a few outcomes, determined for the most part by means of the reaction of the mass immune system. The effectiveness of this reaction is precious by essential factors such as the genetics of the immune system over and above extrinsic factors, e.g., invective to the immune system and the nutritional and physiological state of the host. Innovative drugs and vaccines are essential to stem the worldwide epidemic of tuberculosis so as to kills near about two million people each year. Towards reasonably develop novel anti-tubercular agents, it is essential to study the genetics and physiology of M. tuberculosis and related mycobacteria. It is equally important to understand the M. tuberculosis-host interaction to learn how these bacteria circumvent host defenses and cause disease. The generally it used diagnostic processes for tuberculosis are the tuberculin skin test, acid-fast stain, and chest radiographs. The H37Rv genome strain was published in 1998. With its size of 4 million base pairs and 3959 genes; 40% of these genes have had their function characterised, with possible function postulated for another 44%, within the genome are also six pseudogenes [1].
In fatty acid metabolism process there are near about 250 genes are take a part, in which 39 of these involved in the polyketide metabolism generating the waxy coat. Such huge numbers of conserved genes illustrate the evolutionary significance of the waxy coat to pathogen survival. Additionally, experimental studies have since validated the significance of a lipid metabolism process for M. tuberculosis, consisting exclusively of host-derived lipids for instance fats and cholesterol. M. tuberculosis can furthermore cultivate on the lipid cholesterol as an individual basis of genes and carbon occupied in the cholesterol exploit pathway(s) have been validated because significant throughout different stages of the infection lifecycle of M. tuberculosis, above all throughout the chronic infection phase while additional nutrients are probable not available [2]. M. tuberculosis is a causative representative thus for the reason that to causes tuberculosis and leads to lesions in lungs and other organs. Tuberculosis is the second most important causes of passing away in transmittable diseases [3-5]. In every case the objective of these approaches is to restrict the creation of prospective interactions contained by cells through an iterative process of experimentation, data collection as well as computational approaches so as to consequence in network reconstruction [6,7].
Tuberculosis is a universal predicament because every 15 seconds/death from tuberculosis (2 million deaths per year) with 8 million communities develop tuberculosis annually, exclusive of treatment up to 60% populace infected will be dying. Its major rationales were poverty, lack of healthy living conditions and adequate medical care [8-10]. Tuberculosis continues to affect about 30% humanityʼs populace, predominantly in developing countries, despite existence of chemotherapeutic drugs and widespread use of the Mycobacterium bovis BCG (Bacille Calmette-Guérin) vaccine. Effective chemotherapeutic treatment takes long time, it is expensive, and not available to people in various parts of world where needed most. The circumstances are supplementary convoluted by appearance of multidrug-resistant strains. BCG vaccination effectiveness is as well contentious, as it is not be successful to defend adults in opposition to pulmonary tuberculosis. For predicting sub cellular position of eukaryotic, prokaryotic (Gram-negative as well as Gram-positive bacteria) different methods had been developed but no method has been developing for mycobacterium protein, which may correspond to inventory of potent immune-gens of this trepidation pathogen. In this investigation, challenges were complete to increase a method for prediction of sub cellular location of Mycobacterium proteins [11-13].
M. tuberculosis contamination whichever induces otherwise reduces host cell death, depending scheduled the bacterial strain along with the cell micro-environment. There is confirmation signifying a function on behalf of mitochondria in these processes. In contrast, it has been exposed so as to more than a few bacterial proteins are competent to objective mitochondria, playing a critical role in bacterial pathogenesis and modulation of cell death. However, mycobacteria–derived proteins equipped aim host cell mitochondria are less studied [3].
Consequently, it is significant to envisage sub-cellular localization of protein in pathogenic organism similar to Mycobacterium. Normally, existing processes of sub-cellular localization developed for eukaryotic proteins similar to TSSub (Tsinghua subcellular localization software), LOCSVMPSI (Local Support Vector Machine along with the position-specific scoring matrix PSI-BLAST), ESLpred (Prediction of Eukaryotic Sub-cellular localization), Euk-Ploc (Eukaryotic Protein-sub-cellular Location prediction). A replica has been urbanized for predicting four sub-cellular locations of mycobacterium proteins, namely cytoplasmic, integral membrane, secretary and membrane- attached proteins. The aim behind this study was to predict the sub cellular localization of putative proteins of M. tuberculosis H37Rv strain as they might be useful for targeting anti-micro-bacterial drugs [14]. Tuberculosis (TB) has regrettably retained the position, on behalf of number of decades, as being the leading killer among all infectious diseases. The important gene group identified structure an origin for designing experiments to probe their advanced efficient roles and moreover provide as a ready shortlist for identifying drug targets [15,16].

Biological Networks
Biological system contain many cells and molecules which interact through each one previous and thus forms a network (which consist of nodes and directed and undirected edged) called biological network. Subunits so as to be linked together into a whole in a system are known a network similar to protein-protein interaction network, metabolic network, gene regulatory network. As life is a dynamic process, the study of modelling, analysis and visualisation of biological network are the crucial area of study in life sciences nowadays as biological network is highly structural network. There are several dissimilar types of networks in biology some of which we will be describing briefly.
Protein-protein interaction (PPI) network
One of the on the whole significant types of interaction in biology is protein-protein interaction (figures 1 and 2) which involves the binding of two or more proteins so as to carry out their biological function. Majority of the cellular events such as signal transduction which involves protein–protein interactions for the transport of signals from the extrinsic part of the cells to internal part of the cell and over and above transcriptional regulation which is also based on the interactions of the different proteins, this indicates so as to for the study of biological systems the understanding of interactions at cellular level is needed.
There are different process to investigate the proteinprotein interactions such as high throughput detection method which includes yeast, two hybrid screening and affinity mass spectrometry. Nowadays there are many softwareʼs available for the visualization of protein-protein interaction one of them is cytoscape, which is widely used application for visualization and also for analysis. The biological database has been created where these interactions are collected and preserved together for further study similar to STRING, DIP (Database of Interacting Protein) etc. In a Graphical representation, a node corresponds in the direction of a protein and two proteins have an edge if they physically interact.

Few extremely associated nodes (hubs) embrace the network together. The node color indicate the phenotypic outcome deriving as of removing the corresponding protein, 1. red: lethal, 2. green: non–lethal, 3. orange: slow growth, 4. yellow: unknown.
Materials and Methods
Databases used
1. Super Targets
2. NCBI (National Center for Biotechnology Information) (www.ncbi.nlm.nih.gov/pmc/), (http://www.ncbi.nlm.nih.gov/pubmed).
3. String (Search Tool intended for the Retrieval of Interacting Genes/Proteins)
Super targets: It is a first place developed database used to collect information about drug-target relations. It consists primarily of three dissimilar types of entities:
1. Drugs
2. Proteins
3. Side-Effects
Beside this, information concerning ontologies and pathways is able to be gained. Undo segment is committed to an individual target subgroup-the cytochromes (CYPs) P450. All these entities are associated among every one with drug-protein, protein-protein and drug-side effect associations and embrace prosperous annotation concerning the source, IDʼs, physical properties, references and much more. Proteins are retrieved from UniProt and are displayed with synonyms and organism information. As well in sequence with reference to target-target interactions and sequence similarities among the targets are concerned. Additional information as 3D-structures from PDB and Electronic Configurationnumbers are provided if available. Drugs gained from Super Drug were mapped with BindingDB, integrated to SUPERTARGET and assign metadata as Anatomical Therapeutic Chemical (ATC) codes, arrangement in sequence and binding affinities. The bad-impact of drugs contained in the database was fetch from SIDER and associated to the drugs. The endeavour of SUPERTARGET is to recommend at for the most part comprehensive datasets. Here for target of drug relations from different well-known databases as DrugBank, BindingDB and SuperCyp were incorporated. To improve the entirety of the dataset supplementary novel explored associations were incorporated. The novel-information was obtained in two different steps:
1. Text-mining algorithms were practical to sort all PubMed listed papers by their significance for drugtarget associations.
2. In a second step, the 7,000 papers with the uppermost rank were physically revised.
Protein-protein interaction (PPI) data was obtained from Consensus PathDB which integrates physical protein-protein interactions, metabolic and signaling reactions and gene regulatory interactions (Figure 3). Information on complex composition comes from Corum a protein complex database.
NCBI (national center for Biotechnology Information): It is primary database relevant to biotechnology and biomedicine of the United States National Library of Medicine (NLM), a division of the National Institutes of Health with foremost databases include GenBank for DNA sequences and PubMed, a bibliographic database for the biomedical literature. All these databases are available online through the Entrez search engine. NCBI is directed by David Lipman, one of the original authors of the BLAST sequence alignment program and a widely respected figure in bioinformatics.
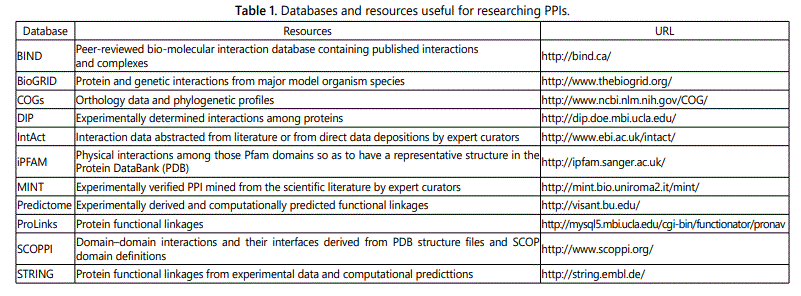
PubChem: PubChem is a database of chemical molecules and their activities against biological assays. The system is maintained by the National Centre for Biotechnology Information (NCBI), a component of the National Library of Medicine, which is part of the United States National Institutes of Health (NIH). PubChem is made up of “three linked databases within the NCBIʼs Entrez information retrieval system. PubChem Substance - “Search deposited chemical substance records using name, synonym or keywords. Links to biological property information and depositor web sites are provided. PubChem Compound - “Search unique chemical structures using names, synonyms or keywords. Links to available biological property information are provided for each compound. PubChem BioAssay - “Search bioassay records using terms from the bioassay description, for example ‘cancer cell line.’ Links to active compounds and bioassay results are provided (Table 1).
String: String is a database of known and predicted protein interaction. The interactions include direct (physical) and indirect (functional) associations; they are derived from four sources:
- Genomic Context.
- High-throughput experiments.
- (Conserved) coexpression.
- Previous knowledge.
To specify your desired starting point of the analysis you have to use the input form at the STRING start page (depicted above). You can enter your protein of interest by supplying its name or identifier. Alternatively, clicking on the other tabs, you can search by amino acid sequence (in any format), multiple names or multiple sequences. There are also 3 example inputs and a random input generator which will randomly select a protein with at least 4 predicted links at medium confidence or better. The organism can be selected by clicking on the arrow or directly typing the name inside the relative input field (an autocompletion mechanism will appear to help you). General names so as to group more than one organism (e.g. “mammals”) can also be used. The network view summarizes the network of predicted associations for a particular group of proteins. The network nodes are proteins. Hovering over a node will display its annotation, clicking on a node gives several details about the protein. The edges represent the predicted functional associations. An edge may be drawn with up to 7 differently colored lines-these lines represent the existence of the seven types of evidence used in predicting the associations. A red line indicates the presence of fusion evidence; a green line-neighborhood evidence; a blue line-co-ocurrence evidence; a purple line-experimental evidence; a yellow line-text mining evidence; a light blue linedatabase evidence; a black line-co-expression evidence. Hovering over an edge will display the combined association score, clicking on it gives you the detailed evidence breakdown.
If predicted associations for your protein are found, they are displayed in a summary view, located just below the view of the network. At the top of the summary your input is shown. If your input gene is a fusion of two functions, both will be shown. Predicted associations are shown immediately below your input, sorted by score. Clicking on the score bullets gives you a breakdown of the individual prediction method scores. Clicking on a gene name gives you the protein sequence over and above a list of similar proteins in STRING. Initially, only predictions with medium (or better) confidence, limited to the top 10 will be shown. These settings can be changed by using the parameter dialog box at the bottom of the page.
Each ‘view’ has a designated set of parameters. The first parameters are the same for all views: Your input identifier, your requested minimum confidence and an option to limit the output to the 10 best-scoring hits. The confidence score is the approximate probability so as to a predicted link exists among two enzymes in the same metabolic map in the KEGG database. Confidence limits are as follows: low confidence-20% (or better), medium confidence-50%, high confidence-75%, highest confidence-95%. Please note so as to parameters are only changed when you press the ‘Update Parameters’ button. The dialogue box shown above is the one for the Network View. Network specific parameters are: ‘edge scaling factor’-this reduces the length of high-scoring edges so that the images will be drawn more compact and low scoring hits will be spread out further. Lower values mean more compact images, higher values will cause more spread. The second parameter is the ‘network depth’. A value higher than 1 means so as to the search for interactions is iterative-after a first round all nodes are themselves again input for a next round of searches. Nodes of a higher iteration will be colored white. Please note so as to this can result in fairly large images so as to may take a while to compute and download. This feature allows you to ‘walk’ through the network of functional associations. Note so as to you can click on any node, and the subsequent page offers a link to use so as to node as the input-effectively placing it in the center of the image. Repeated use of this mechanism allows you to explore large regions of the network.

Software used
Software: Cytoscape is a general-purpose, open-source software environment for the large scale integration of molecular interaction network data. The Cytoscape Core handles basic features such as network layout and mapping of data attributes to visual display properties. Cytoscape was developed at the Institute of System Biology in Seattle in 2002. It was made publically in July 2002 (V0.8). It is written in java and used in any java-based operating systems. There are many additional plugin available for network and molecular profiling analysis, novel layouts, additional file format support and connection with databases and searching in large networks [17,18]. The software cytoscape V-2.8.3 was downloaded from www.cytoscape.org as an open source java application as it runs on all major operating systems. This software was used for integrating, visualizing, analyzing data in the context of networks (Figure 4). Access to large amount of molecular interaction data is available through different database. However it is difficult to integrate interactions from these data bases so so as to the resulting molecular networks can be visualized and analyzed. Plug-in module of cytoscape provides means to implement the novel algorithms, additional network analysis and biological semantics. Plug-ins is given access to the core network model and can also control the network display [19].
System requirements: The system requirements for Cytoscape depend on the size of the networks the user wants to load, view and manipulate (Table 2).

Result and Discussion
Construction of biological network models
A set of network connections is a set of items, which we will call vertices or sometimes nodes, with connections among them, called edges (Figure 5). Systems taking the form of networks (also called “graphs” in much of the mathematical literature) abound in the world. Examples include the Internet, the World Wide Web, social networks of acquaintance or other connections among individuals, organizational networks and networks of business relations among companies, neural networks, metabolic networks, food webs, distribution networks such as blood vessels or postal delivery routes, networks of citations among papers, and many others.

Degree distribution
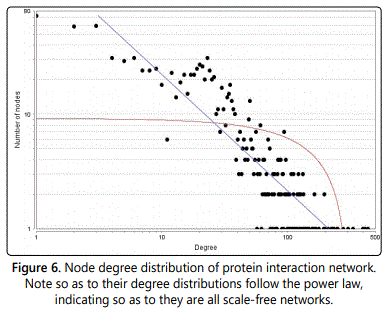
In undirected networks, the node degree of a node n is the number of edges linked to n. A self-loop of a node is counted similar to two edges for the node degree. The node degree distribution gives the number of nodes with degree k for k=0,1, ... In directed networks, the in-degree of a node n is the number of incoming edges and the out-degree is the number of outgoing edges. Similar to undirected networks, there are an in-degree distribution and an out-degree distribution (Figure 6).
Clustering coefficients
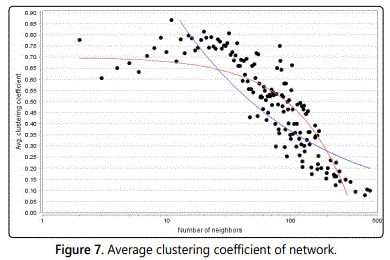
In undirected networks, the clustering coefficient Cn of a node n is defined as Cn= 2en /kn(kn-1), where kn is the number of neighbours of n and en is the number of connected pairs among all neighbours of n. In directed networks, the definition is slightly different: Cn= en/kn(kn-1). In both cases, the clustering coefficient is a ratio N/M, where N is the number of edges among the neighbours of n, and M is the maximum number of edges so as to could possibly exist among the neighbours of n. The clustering coefficient of a node is always a number among 0 and 1.
The average clustering coefficient distribution gives the average of the clustering coefficients for all nodes n with k neighbours for k=2, ... Network Analyzer also computes the network clustering coefficient so that is the average of the clustering coefficients for all nodes in the network. The clustering coefficient of a node is the number of triangles (3-loops) so as to pass through this node, relative to the maximum number of 3-loops that could pass through the node. We didnʼt find any type of clustering coefficient in various types of ER graph model and Barabasi Albert graph, inside the Gephi and also for Barabasi Albert graphs in Cytoscape (Figure 7).
Shortest paths
The length of the shortest path among two nodes n and m is L(n,m). The shortest path length distribution gives the number of node pairs (n,m) with L(n,m)=k for k=1,2,.... The network diameter is the maximum length of shortest paths among two nodes. If a network is disconnected, its diameter is the maximum of all diameters of its connected components. The network diameter and the shortest path length distribution may indicate small-world properties of the analyzed network (Figure 8).

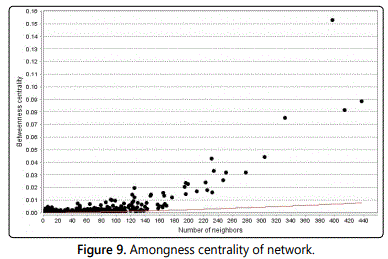
Amongness and closeness centrality
Amongness centrality: Amongness centrality is computed only for networks so as to do not contain multiple edges. The amongness value for each node n is normalized by dividing by the number of node pairs excluding n: (N-1)(N-2)/2, where N is the total number of nodes in the connected component so as to n belongs to. Thus, the amongness centrality of each node is a number among 0 and 1.
The amongness centrality of a node reflects the amount of control so as to this node exerts over the interactions of other nodes in the network. This measure favours nodes so as to join communities (dense sub networks), rather than nodes so as to lie inside a community (Figure 9).
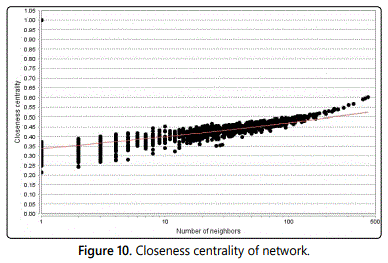
Closeness centrality: The closeness centrality Cn(n) of a node n is defined as the reciprocal of the average shortest path length and is computed as follows:
Cn(n)= 1/avg(L(n,m)) Where L(n,m) is the length of the shortest path among two nodes n and m. The closeness centrality of each node is a number among 0 and 1. Closeness centrality is a measure of how fast information spreads from a given node to other reachable nodes in the network (Figure 10).
Avg. clustering coefficient
As an alternative to the global clustering coefficient, the overall level of clustering in a network is measured by Watts and Strogatz as the average of the local clustering coefficients of all the vertices n:
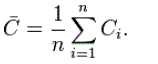
It is worth noting so as to this metric places more weight on the low degree nodes, while the transitivity ratio places more weight on the high degree nodes. In fact, a weighted average where each local clustering score is weighted by ki(ki-1) is identical to the global clustering coefficient.
A graph is considered small-world, if its average local clustering coefficient C is significantly higher than a random graph constructed on the same vertex set, and if the graph has approximately the same mean-shortest path length as its corresponding random graph.
The networks with the largest possible average clustering coefficient are found to have a modular structure, and at the same time, they have the smallest possible average distance among the different nodes (Figure 11).

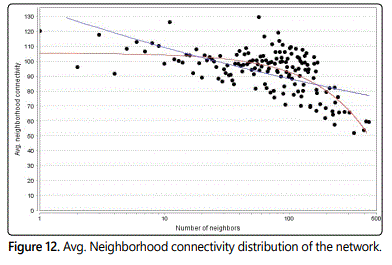
Avg neighborhood connectivity
The connectivity of a node is the number of its neighbors. The neighborhood connectivity of a node n is defined as the average connectivity of all neighbors of n. The neighborhood connectivity distribution gives the average of the neighborhood connectivities of all nodes n with k neighbors for k=0,1,…. Network Analyzer computes similar parameters for directed networks. In analogy to the in-and out-degree, every node n in a directed network has in- and outconnectivity. Thus, in directed networks, a node has the following types of neighborhood connectivity:
- only in-the average out-connectivity of all in-neighbors of n;
- only out-the average in-connectivity of all out-neighbors of n;
- in and out-the average connectivity of all neighbors of n (edge direction is ignored).
Based on the three definitions given above, there are three neighborhood connectivity distributions-“only in”, “only out” and “in and out”.
If the neighborhood connectivity distribution is a decreasing function in k, edges among low connected and highly connected nodes prevail in the network (Figure 12).
Shared neighbor
P(n,m) is the number of partners shared among the nodes n and m, so as to is, nodes so as to are neighbors of both n and m. The shared neighbors distribution gives the number of node pairs (n,m) with P(n,m)=k for k=1,….
If a motif similar to the one presented in figure 5 is over-represented in a network, this can be inferred from the shared neighbors distribution (Figure 13).
Stress centrality
The stress centrality of a node n is the number of shortest paths passing through n. A node has a high stress if it is traversed by a high number of shortest paths. This parameter is defined only for networks without multiple edges (Figure 14).
The stress centraility distribution gives the number of nodes with stress s for different values of s. The values for the stress are grouped into bins whose size grows exponentially by a factor of 10. The bins used for this distribution are {0}; (1, 10); (10, 100); ...

Topological coefficient
The topological coefficient Tn of a node n with kn neighbors is computed as follows:
Tn=avg (J(n,m))/kn .
Here, J(n,m) is defined for all nodes m so as to share at least one neighbor with n. The value J(n,m) is the number of neighbors shared among the nodes n and m, plus one if there is a direct link among n and m.
For example, J(b,c)=J(b,d)=J(b,e)=2. Therefore, Tb=2/3.
The topological coefficient is a relative measure for the extent to which a node shares neighbours with other nodes (Table 3). Network Analyzer computes the topological coefficients for all nodes with more than one neighbor in the network. Nodes so as to have one or no neighbors are assigned a topological coefficient of 0 (zero). The chart of the topological coefficients can be used to estimate the tendency of the nodes in the network to have shared neighbors (Figure 15).


Hubs
In a scale-free network, small-degree nodes are the most abundant, but the frequency of high-degree nodes decreases relatively slowly (Figure 16). Thus, nodes so as to have degrees much higher than average, so-called hubs exist. Because of the heterogeneity of scale-free networks, random node disruptions do not lead to a major loss of connectivity, but the loss of the hubs causes the breakdown of the network into isolated clusters (Figure 17).

Characterization of hubs

Conclusion
PPI networks make available an easy general idea of the network of communications consequently at the same time as to obtain consign inside a cell. The immense amounts of sequence data quantities consequently as to have been generated. It also has been leveraged to construct enhanced predictions of communications and functional links between proteins, over and above individual protein functions. By integrating investigational process intended for influential PPIs and computational methods for prophecy, very many constructive and functional data on PPIs have been generated, together with a number of far above the ground eminence databases.
Acknowledgement
Person responsible would similar to express gratitude Dr Prashant Ankur Jain, Assistant Professor and In-charge, Department of Computational Biology and Bioinformatics, Jacob Institute of Biotechnology and Bioengineering, Sam Higginbottom University of Agriculture, Technology and Sciences (SHUATS), Allahabad, U.P.-India and Dr Satyam Khanna, Managing Director, RASS Bio-solution PVT. Ltd., Civil-Line, Kanpur, Uttar Pradesh (U.P.)-India, for arranging a vigorous & reasonable research ambience.
References
- Ryan KJ, Ray CG, Sherries JC. Sherris Medical Microbiology: An Introduction to Infectious Diseases. 4th edition. Novel York: McGraw-hill; 2004.
- Wipperman MF, Sampson NS, Thomas ST. Pathogen Roid Rage: Cholesterol Utilization by Mycobacterium tuberculosis. Crit Rev Biochem Mol Biol. 2014; 49(4): 269-293. doi:10.3109/10409238.2014.895700
- Moreno-Altamirano MM, Paredes-González IS, Espitia C, SantiagoMaldonado M, Hernández-Pando R, Sánchez-García FJ. Bioinformatic Identification of Mycobacterium tuberculosis Proteins likely to Target Host Cell Mitochondria: Virulence Factors? Microb Inform Exp. 2012; 2(1): 9. doi: 10.1186/2042-5783-2-9.
- Fang X, Wallqvist A, Reifman J. Modeling Phenotypic Metabolic Adaptations of Mycobacterium tuberculosis H37Rv under Hypoxia. PLoS Comput Biol. 2012; 8(9): e1002688. doi:10.1371/journal.pcbi.1002688.
- Liu ZP, Wang J, Qiu YU, et al. Inferring Protein-Protein Interactions Based on Sequences and Interologs in Mycobacterium Tuberculosis. In: Huang DS, Gan Y, Premaratne P, Han K (eds). Bio-Inspired Computing and Applications. ICIC 2011. Lecture Notes in Computer Science. Springer, Berlin, Heidelberg. 2012; 6840: 91-96. doi: 10.1007/978-3-642-24553-4_14
- Minch KJ, Rustad TR, Peterson EJ, et al. The DNA-Binding Network of Mycobacterium Tuberculosis. Nat Commun. 2015; 6: 5829. doi: 10.1038/ncomms6829.
- Parish T, Brown AC. Mycobacterium: Genomics and Molecular Biology. 1st Edition. Norfolk UK: Caister Academic Press; 2009.
- Van Soolingen D. Molecular Epidemiology of Tuberculosis and Other Mycobacterial Infections: Main Methodologies and Achievements. J Intern Med. 2001; 249(1): 1-26. doi: 10.1046/j.1365-2796.2001.00772.x.
- Frothingham R, Meeker-OʼConnell WA. Genetic Diversity in the Mycobacterium Tuberculosis Complex Based on Variable Numbers of Tandem DNA Repeats. Microbiology. 1998; 144(5): 1189-1196. doi: 10.1099/00221287-144-5-1189
- Mazars E, Lesjean S, Banuls AL, et al. High-Resolution Minisatellite-Based Typing as a Portable Approach to Global Analysis of Mycobacterium Tuberculosis Molecular Epidemiology. Proc Natl Acad Sci U S A. 2001; 98(4): 1901-1906, doi: 10.1073/pnas.98.4.1901.
- Hawkey PM, Smith EG, Evans JT, et al. Mycobacterial interspersed repetitive unit typing of Mycobacterium tuberculosis compared to IS6110-based restriction fragment length polymorphism analysis for investigation of apparently clustered cases of tuberculosis. J Clin Microbiol. 2003; 41(8): 3514-3520.
- Supply P, Allix C, Lesjean S, et al. Proposal for Standardization of Optimized Mycobacterial Interspersed Repetitive Unit-Variable-Number Tandem Repeat Typing of Mycobacterium Tuberculosis. J Clin Microbiol. 2006; 44(12): 4498-4510. doi: 10.1128/JCM.01392-06.
- Müller R, Roberts CA, Brown TA. Complications in the Study of Ancient Tuberculosis: Non-Specificity of IS6110 PCRs. STAR: Science and Technology of Archaeological Research. 2015; 1(1): 1-8. doi: 10.1179/2054892314Y.0000000002.
- Gomez M, Johnson S, Gennaro ML. Identification of Secreted Proteins of Mycobacterium tuberculosis by a Bioinformatic Approach. Infection and Immunity. 2000; 68(4): 2323-2327. doi: 10.1128/IAI.68.4.2323-2327.2000
- Goldman RC, Plumley KV, Laughon BE. The Evolution of Extensively Drug Resistant Tuberculosis (XDR-TB): History, Status and Issues for Global Control. Infect Disord Drug Targets. 2007; 7(2): 73-91. doi: 10.2174/187152607781001844.
- Ghosh S, Baloni P, Mukherjee S, Anand P, Chandra N. A Multi-Level MultiScale Approach to Study Essential Genes in Mycobacterium Tuberculosis. BMC Syst Biol. 2013; 7: 132. doi: 10.1186/1752-0509-7-132.
- Bell GW, Lewitter F. Visualizing networks. Methods Enzymol. 2006; 411: 408-421. doi: 10.1016|50076-6879(06)11022-8
- Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003; 13(11): 2498-2504. doi: 10.1101|gr|239303
- Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci USA. 2001; 98(8): 4569-4574.
