

Research Article
TLR3 Deficiency Leads to a Dysregulation in the Global Gene-Expression Profile in Murine Oviduct Epithelial Cells Infected with Chlamydia muridarum
Department of Microbiology and Immunology, Indiana University School of Medicine, Indianapolis, Indiana-46202, USA
*Corresponding author: Wilbert A Derbigny, Assistant Research Professor, Department of Microbiology and immunology, Indiana University School of Medicine, 635 Barnhill Drive, Indianapolis, Indiana, USA, Tel: +1 (317) 274-0697, Fax: (317) 274-4090, E-mail: wderbign@iupui.edu
Received: November 14, 2018 Accepted: November 28, 2018 Published: December 5, 2018
Citation: Kumar R, Derbigny WA. TLR3 Deficiency Leads to a Dysregulation in the Global Gene-Expression Profile in Murine Oviduct Epithelial Cells Infected with Chlamydia muridarum. Int J Microbiol Curr Res. 2018; 1(1): 1-13. doi: 10.18689/ijmr-1000101
Copyright: © 2018 The Author(s). This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Chlamydia trachomatis replicates primarily in the epithelial cells lining the genital tract and induces the innate immune response by triggering cellular pathogen recognition receptors (PRRs). Our previous studies showed that Toll-like receptor 3 (TLR3) is expressed in murine oviduct epithelial (OE) cells, is the primary PRR triggered by C. muridarum (Cm) early during infection to induce IFN-β synthesis, and that TLR3 signaling regulates the chlamydial induced synthesis of a plethora of other innate inflammatory modulators including IL-6, CXCL10, CXCL16 and CCL5. We also showed that the expression of these cytokines induced by Chlamydia was severely diminished during TLR3 deficiency; however, the replication of Chlamydiain TLR3 deficient OE cells was more robust than in WT cells. These data suggested that TLR3 had a biological impact on the inflammatory response to Chlamydia infection; however, the global effects of TLR3 signaling in the cellular response to Chlamydia infection in murine OE cells has not yet been investigated. To determine the impact of TLR3 signaling on Chlamydia infection in OE cell at the transcriptome level, we infected wild-type (OE-WT) and TLR3-deficient (OE-TLR3KO) cells with Cm, and performed transcriptome analyses using microarray. Genome-wide expression and ingenuity pathway analysis (IPA) identified enhanced expression of host genes encoding for components found in multiple cellular processes encompassing: (1) pro-inflammatory, (2) cell adhesion, (3) chemoattraction, (4) cellular matrix and small molecule transport, (5) apoptosis, and (6) antigen-processing and presentation. These results support a role for TLR3 in modulating the host cellular responses to Cm infection that extend beyond inflammation and fibrosis, and shows that TLR3 could serve a potential therapeutic target for drug and/or vaccine development.
Keywords: TLR3; Chlamydia; RNA; Micro-array; Epithelial; RANTES; Pathogenesis; Transcriptome; Genital tract.
Introduction
Chlamydia trachomatis is a gram-negative, obligate intracellular bacterial pathogen causing the most common sexually transmitted infections (STI) worldwide, particularly among young women [1]. In women, chronic infection with the urogenital serovars (D-K) can cause pelvic inflammatory diseases (PID) and chronic pelvic pain, which can culminate into scarring and fibrosis of the Fallopian tubes leading to infertility or ectopic pregnancy [2-4]. According to the Centers for Disease Control and Preventionʼs (CDCʼs) report in 2016, a total of 1,598,354 Chlamydia infections were reported in USA alone, which was 4.7% higher than the reported number of cases in 2015 [5]. As per CDCʼs estimate, nearly 20 million healthy individuals are infected every year, accounting for almost $16 billion in health care costs annually (CDC 2016) [5].The asymptomatic nature of C. trachomatis infection means that the pathogen persists for a long time in some individuals, which suggests the effective evasion of host immune systems [6,7].
The innate immune system is known for recognizing a vast variety of pathogens, including viruses, bacteria and fungi via sensing the specific pathogen associated molecular patterns (PAMPs). The innate immune response induced by PAMPs includes the cellular production of a wide range of antimicrobial and inflammatory mediators. The recognition of PAMPs by pattern recognition receptors (PRRs) expressed by innate immune cells is crucial for maintenance of homeostatic immunity as well as an effective induction of an adaptive immune response [8-10]. However, an overly activated innate immune response can cause the overabundant production of inflammatory mediators which can result in tissue damage [11,12]. Among the PRRs, Toll-like receptors (TLRs) play a major role in innate immunity by recognizing structurally conserved microbial components [13]. TLRs are membrane bound PRRs that have been shown to be triggered by PAMPs from various bacterial, viral and fungal pathogens [14]. Engagement of the TLRs by the PAMPs can lead to the activation of phagocytosis and the production of inflammatory cytokines including TNF-α, IL-6, and GMCSF, as an important step prior to the switch from an innate immunity and the onset of an adaptive immune response [15-18].
Epithelial cells lining the mucosal surfaces of the female genital tract serve as the sentinels to the invading C. trachomatis by expressing TLRs that trigger the innate immune response by inducing a multitude of pro-inflammatory cytokines and chemokines during infection [19]. These cytokines and chemokines mediate resolution of Chlamydia infection and are responsible for polarizing the innate and adaptive immune responses [18-23]. We previously showed that Chlamydia muridarum (Cm), an orthologue of human C. trachomatis, induces TLR2 dependent secretion of acute-phase inflammatory cytokines including IL-6, GM-CSF, and TNF-α in murine oviduct epithelial (OE) cells [8]. Further, we showed that Chlamydia infection mediates IFN-β secretion in a mostly TLR3-dependent manner, and demonstrated a role for TLR3 in regulating the expression of a plethora of other innate-inflammatory modulators including IL-6, CXCL10, CXCL16 and CCL5 [24-26]. The Chlamydia-induced expression of these cytokines was severely diminished in TLR3 deficient OE cells whereas; replication of Cm in TLR3 deficient OE cells was more robust than in WT OE cells. These findings suggest that triggering the TLR3 pathway in OE cells during Cm infection invokes cellular mechanisms that inhibit the Chlamydial developmental cycle, and thereby implicate TLR3 in regulating cellular processes that extend beyond the syntheses of chemo tactic and inflammatory mediators.
In order to help identify other cellular pathways that are regulated by TLR3 signaling during Chlamydia infection, we conducted transcriptome analyses and comparative gene expression profiling on WT and TLR3-deficient murine OE cells that were infected with Cm. In the present study, we aim to identify target genes that are affected by Chlamydia infection of OE cells and to ascertain a role for TLR3 in regulating those pathways by using genome-wide microarray analysis followed by ingenuity pathway analysis (IPA). Our results indicate that TLR3 plays a significant role in modulating the host cellular responses that encompass a diverse subset of biological functions during Cm infection.
Materials and Methods
Oviduct epithelial cells and culture conditions
The murine oviduct epithelial cell lines OE-129TLR3-/- (C19) and OE-129WT [25] were grown at 37°C in a 5% CO2 humidified incubator in epithelial-cell media {Dulbeccoʼs modified Eagle medium and F12K (Sigma-Aldrich) in 1:1 ratio}, supplemented with 10% fetal bovine serum (HyClone), 2mM L-alanyl-L-glutamine (Glutamax I; Gibco/ Invitrogen, Carlsbad, CA), 5µg bovine insulin/ml, and 12.5 ηg/ml recombinant human KGF (keratinocyte growth factor; Sigma-Aldrich) as previously described [18,24].
Chlamydia stocks
Mycoplasma-free C. muridarum Nigg, previously known as C. trachomatis strain (MoPn), was grown and titrated in McCoy cells (ATCC) as described [18, 27, 28]. The elementary bodies were harvested from infected McCoy cells, were re-suspended in SPG buffer (250mM sucrose, 10 mM sodium phosphate, and 5mM L-glutamic acid, pH 7.2), and quantified on McCoy cells using methodology described previously [18,26,29].
In vitro infection of oviduct epithelial cells
OE-129WT and OE-129TLR3-/- (C19) cells were seeded in 6-well tissue culture plates and grown until confluence. The cells were infected with 5 inclusion forming-units (IFU) of C. muridarum/cell in 900ul of culture medium as described previously [24, 25]. Mock-treated controls were incubated with an inoculum containing equivalent volume of SPG buffer, but were not infected with Chlamydia. Each cell type was seeded in duplicate wells of the 6-well plate and were infected when confluent. The experiments were repeated 3 times on different days to provide the appropriate number of biological replicates for proper statistical analyses.
Total RNA purification and Microarray analysis
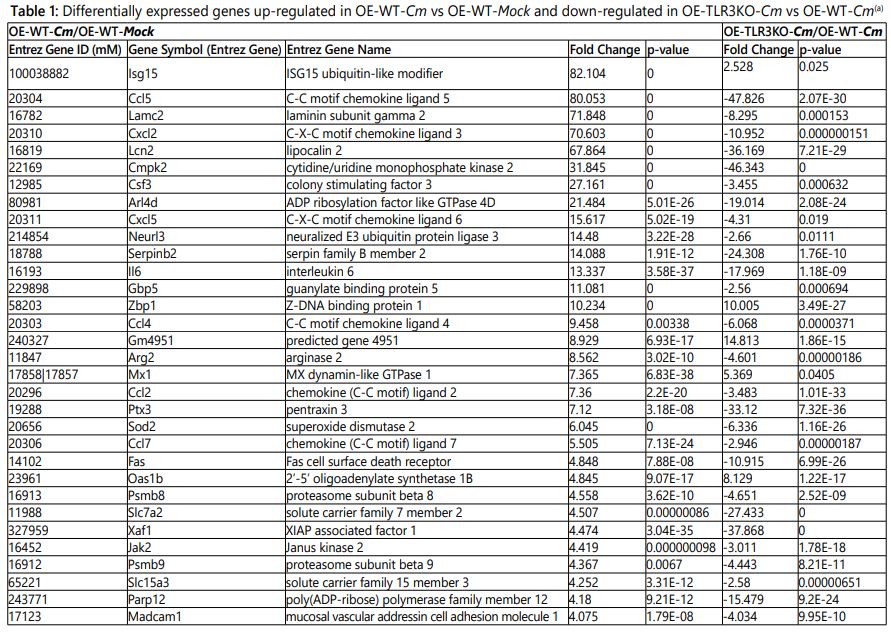
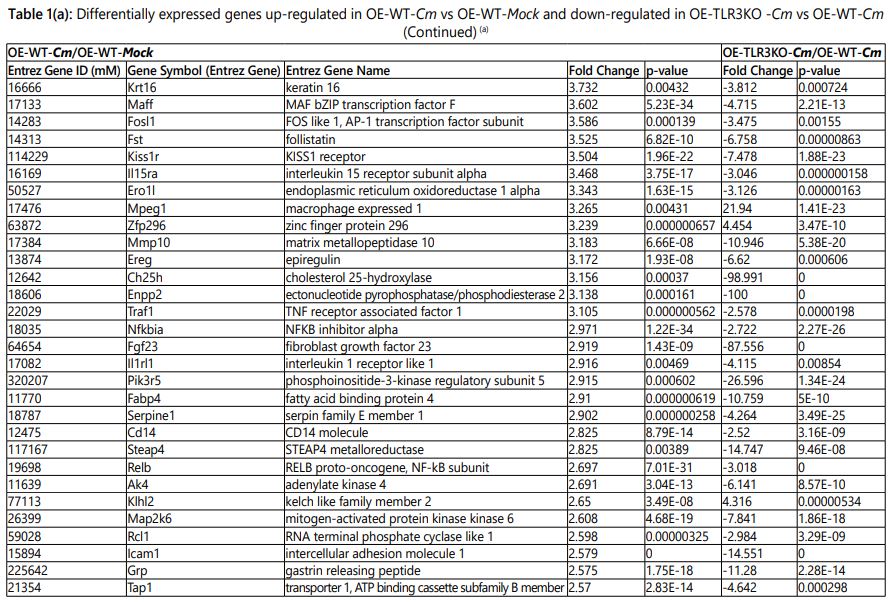
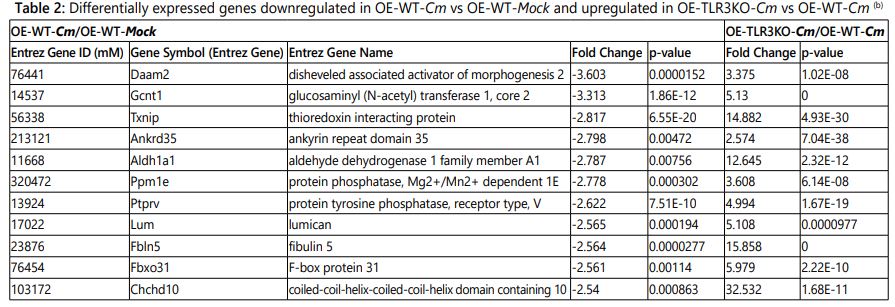
Total RNA was purified from OE-129WT and OE-129TLR3-/-(C19) cells infected with C. muridarum(5 IFU/cell) at 24h post-infection using the Norgen Total RNA Purification Kit(Norgen Biotek; Thorold, Ontario). During purification, all RNA samples were treated with RNase-free DNase I (Qiagen) to remove all traces of genomic-DNA contamination. The RNA quantity and quality were measured using the Nano Drop 2000c spectrophotometer (Thermo Scientific; Pittsburgh, PA). The RNA purified from mock-infected cells was used as control sample. Purified total RNA was submitted to the gene-expression profiling services of Phalanx Biotech Group, Inc. (Belmont, CA) for microarray analysis. Genes that were up or down regulated ≥ 2.5-fold with p-values <0.05 were considered in the final analysis (Tables 1 and 2; and Supplementary Tables 1 and 2).
Functional and canonical pathway analyses
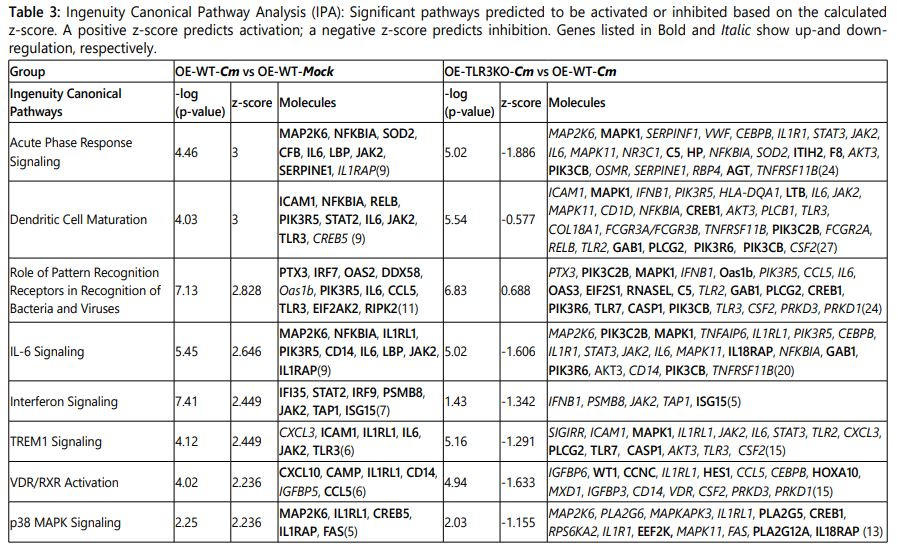
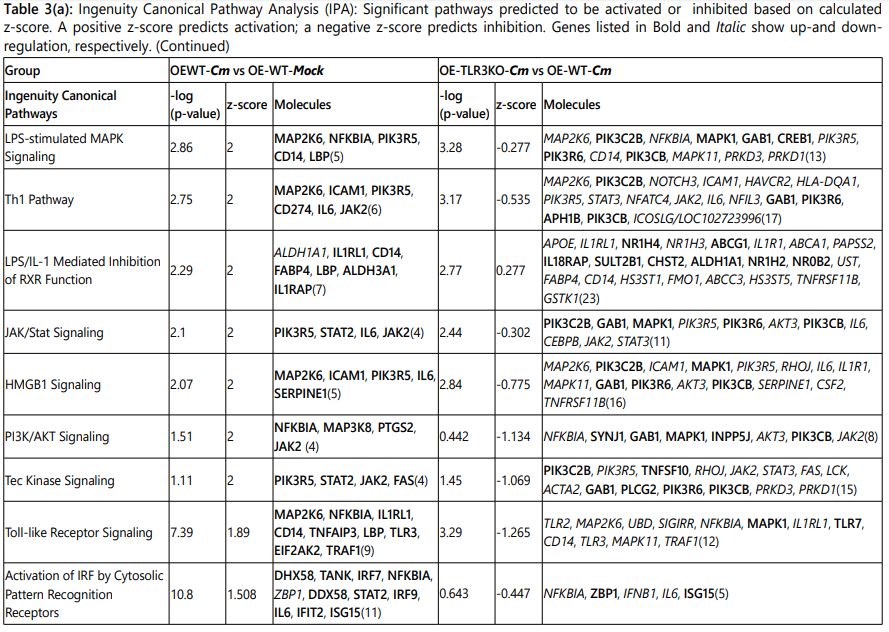
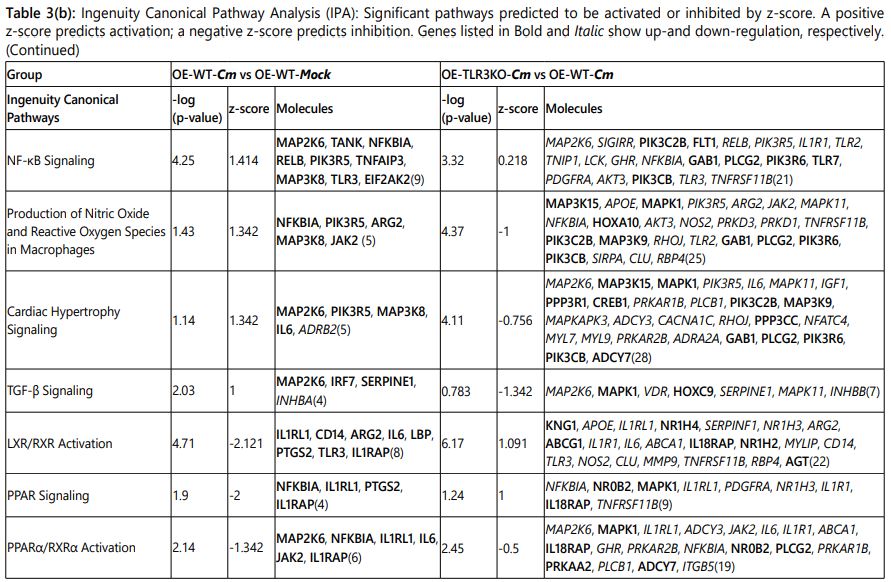
The microarray gene expression data were analyzed by Qiagenʼs ingenuity pathway analysis (IPA; Ingenuity Systems, https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis; Qiagen Inc.) to determine whether genes are associated with particular diseases, exhibit prominent biological function, or if canonical signaling pathways were preferentially up- or down-regulated in murine OE cells [30]. Genes were selected for analyses if they had a p-value <0.05 and absolute fold change ≥2.5. The data are presented as the comparison of: (1) C. muridarum-infected wild-type (OE-WT-Cm)vs Mock-infected wild-type (OE-WT-Mock) and (2)C. muridarum-infected wild-type (OE-WT-Cm)vs C. muridarum-infected TLR3-deficient OE cells (OE-TLR3KO-Cm) (Table 3, Supplementary Table 3, and Table 4).
Results
Identification of differentially-regulated genes in murine oviduct epithelial cells
Our previous studies showed that TLR3 is the primary PRR in OE cells triggered by Cm infection in the early synthesis of IFN-β, and in the syntheses of a multitude of other innate inflammatory modulators such as IL-6, CXCL10, CXCL16 and CCL5. Our data show that TLR3 has a biological impact on the innate immune response to Chlamydia infection; however, the comprehensive impact of TLR3 deficiency during Chlamydia infection of murine OE cells remains unclear. To determine the global significance of TLR3 signaling on Cm infection in OE cell lines, we infected wild-type and TLR3-deficient OE cells (here forth referred to as OE-WT and OE-TLR3KO cells, respectively) with 5 IFU/cell Cm for 24h post-infection (PI) and performed transcriptome analysis using microarray. We used a minimum significance criterium of p<0.05 and an absolute fold change ≥2.5 for comparative gene analyses (See Materials and Methods). Out of the several hundred genes that were differentially affected by Cm infection, candidate gene selection based on this criterion yielded 152 genes up-regulated and 56 genes down-regulated when comparing OE-WT-Cm vs the OE-WT-Mock control (Supplementary Tables 1 and 2). Interestingly, there were 600 genes up-regulated and 616 genes down-regulated when comparing OE-TLR3KO-Cm vs OE-WT-Cm cells, which shows that TLR3 deficiency results in the differential regulation of a multitude of cellular processes during Chlamydia infection of the OE cells.
We selected genes that were up-/or down-regulated > 2.5-fold in both comparative groups: [OE-WT-Cm vs OE-WT-Mock] and [OE-TLR3KO-Cm vs OE-WT-Cm] for our analyses (Tables 1 and 2).The most prominent genes up-regulated in OE-WT-Cm vs OE-WT-Mock are listed in Table 1. As indicated, many of the most highly up-regulated genes in the OE-WT-Cm cells such as Ccl5 (80.050-fold), Lamc2 (71.848-fold), Cxcl2 (70.603-flod), Lcn2 (67.864-fold), Cmpk2 (31.845 fold) and Csf3 (27.161-fold) were substantially and significantly down regulated in OE-TLR3KO-Cm cells. Not surprisingly, pro-inflammatory cytokines and chemokines showed the highest level of up regulation during Cm infection in the OE-WT cells. Amongst the most highly up-regulated pro-inflammatory mediators were Ccl5 (RANTES), Ccl4 (MIP-1b), and the monocyte chemo attractant proteins Ccl2 and Ccl7 (or MCP 1 and 3, respectively) belonging to C-C chemokine family. The C-X-C motif chemokines Cxcl2 (also called macrophage inflammatory protein2-alpha) and Cxcl5 (or epithelial neutrophil activating protein-78), and the acute inflammatory cytokines Csf3 and Il6 were also included in the most highly upregulated inflammatory mediators. These highly up-regulated cytokine and chemokines were severely down-regulated in the OE-TLR3KO-Cm cells relative to OE-WT-Cm cells (Table1).
The extracellular matrix (ECM) proteins play a critical role in the cell invasion, adhesion, cell patterning and architectural changes, and “outside-in” signal transduction [31]. Our microarray data showed that many host ECM moieties were differentially expressed, suggesting a rapid and dramatic remodeling of the extracellular milieu in response to Chlamydia infection (Tables 1 and 2; Supplementary Tables 1 and 2). The genes involved in remodeling included glycol proteins, metalloproteinases, numerous collagens, and several fibrosis-associated moieties that were highly upregulated in our data set (Table 1). The Cm induced ECM genes included Lamc2 (LAMC2), Fst (Follistatin), Mmp10 (metalloproteinase MMP10), Ereg (epiregulin EREG), Enpp2 (ATX or autotaxin, ENPP2), Fgf23 (fibroblast growth factor FGF23), Steap4 (metalloreductase STEAP4), Madcam-1(mucosal vascular address in cell adhesion molecule-1, MAdCam-1) and Icam-1 (intracellular adhesion molecule-1, ICAM-1). However, as we have seen with many of the highly up regulated pro-inflammatory mediators, transcription of these ECM genes in the Cm-infected OE-TLR3KO cells resulted in a substantial down regulation or inhibition when compared to the Cm-infected OE-WT cells.
Other genes that were highly up regulated in response to Cm infection in WT-OE cells but attenuated during TLR3 deficiency included components of various cellular metabolic pathways (LCN2, CMPK2, ARG2, SOD2, ERO1-α [or endoplasmic reticulum oxidoreductase1-α], CH25H, FABP4, and AK4), and proteins involved in membrane tracking and protein processing (ARL4D, NEURL3, PTX3, SERPINB2 [or PAI-2], and SERPINE1 [or PAI-1]). Interferon-sensitive genes (ISGs) are known for their role in cell-intrinsic immunity against diverse pathogens, such as viruses and intracellular bacterial species including Chlamydia, Mycobacteria, Listeria, Salmonella, and Toxoplasma [32]. We found several ISG sincluding Gbp5 (a member of IFN-inducible subfamily of GTPases) [33] and Parp12 [34-37] that were highly induced in the Cm infected OE-WT cells, but differentially regulated during TLR3 deficiency.
Some of the more moderately up-regulated genes encoding the pro-apoptotic proteins (FAS and XAF1), proteins related to MHC Class 1 antigen presentation (PSMB-8, PSMB-9 and TAP1), solute carrier (SLC) transporters (SLC7A2 and SLC15A3), and the JAK2 protein [38]. Other genes moderately up regulated included those encoding the TNF-α mediated pro-inflammatory proteins KRT16 (keratin), activator protein-1 (AP-1), family members of MAFF (MafbZIP transcription factor F) and FOSL1 (Fos-like 1or FRA1) [39], TRAF1, NF-κBia (inhibitor of nuclear factor κappa-B or IκB-α), NF-κB/RELB, MKK6 (Mitogen-activated protein kinase 6), and GRP (Gastrin releasing peptide). Finally, our data also showed some moderate but significantly up-regulated expression of genes encoding various receptors and signaling molecules including KISS1R, IL15R-α, IL1RL, CD14, PIK3R5 (phosphatidylinositol 3-kinase, regulatory subunit 5), and the RCL1(RNA terminal phosphate cyclase like-1)protein that is involved in eukaryotic 18S RNA biogenesis [40]. However, much like the highly up regulated genes, these moderately induced genes in Cm-infected OE-WT cells were differentially regulated during TLR3-deficiency. Interestingly, not all genes induced during Cm infection of WT-OE cells were attenuated or down regulated during TLR3 deficiency. Results depicted in Table 1 show that transcription levels for Isg15, Mx1, Gm4951, Oas1b, Mpeg1, Zfp296, Zbp1, and Klhl2 were not reduced in the Cm-infected OE-TLR3KO cells relative to Cm-infected OE-WT cells; instead, their expression levels were significantly increased during TLR3-deficiency. Isg15, Mx1, Oas1b and Mpeg1 are type I and II interferon responsive genes and have been implicated in the host protection against various pathogenic organisms [32, 41-45].
Table 2 shows 11 genes that were down-regulated more than 2.5-fold during Cm infection of the OE-WT cells. Aldh1a1, Ankrd35, Chchd10, Daam2, Fbln5, Fbxo31, Gcnt1, Lum, Ppm1e, Ptprv and Txnip were all down regulated during Cm infection in wild-type OE cells; however, their expression levels in OE-TLR3KO cells was several-fold higher. Interestingly, 8 of the 11 genes (Aldh1a1, Ankrd35, Chchd10, Daam2, Fbln5, Fbxo31, Gcnt1, and Ppm1e) have been reported to be prognostic markers in a variety of metastatic malignancies and other cancer-related diseases [46-54]. Ptprv and Txnip exhibit tumor-suppressor function and has involvement in cell proliferation and apoptosis [55, 56]. The Lum gene encodes the extracellular matrix protein lumican (LUM) that has a role in the bacterial clearance [57, 58], and its expression was upregulated 5.108-fold in Cm-infectedOE-TLR3KO cells when compared to Cm infected OE-WT cells.
Functional and canonical pathway analyses
Based on the gene expression data, a set of cellular path-ways predicted to be either activated or inhibited during Cm infection was identified by IPA software. Table 3 lists what are predicted to be the most significantly changed pathways based on the expression levels of the input genes. As indicated, most of the Cm infection-induced pathways in the OE-WT cells were found to be significantly inhibited in the Cm-infected OE-TLR3KO cells. Pathways regulating the role of PRRs in recognition of bacteria and viruses, LPS/IL-1 mediated inhibition of RXR function, and NF-κB signaling were not inhibited in the Cm-infected OE-TLR3KO cells when compared to Cm-infected WT-OE cells like the other pathways listed in Table 3; however, activation of these pathways was significantly lower in the OE-TLR3KO cells. The lower portion of Table 3 shows the canonical pathways that were predicted to be inactivated in Cm-infected OE-WT cells e.g., LXR/RXR activation, PPAR signaling, and PPARα/RXRα activation pathways. In contrast, LXR/RXR activation and PPAR signaling pathways are predicted to be activated in the Cm-infected OE-TLR3KO cells when compared to the Cm-infected OE-WT cells.
Upstream regulator analysis
IPA predicts upstream transcriptional regulators based on experimentally observed relationships between regulators and genes in the dataset. The calculated z-score predicts either activation or inhibition of regulators on the basis of the relationship with dataset genes and direction of expression change of input genes. There are 7 categories: immunological (cytokines and chemokines), chemical, kinase, transcription factor, trans membrane receptor, translation regulator, and transporter, where several Cm activated and inhibited upstream regulators are grouped together and listed in Supplementary Table 3 (Excel file). Among the 7 regulator categories, cytokine is the top-most significantly activated upstream regulator during Cm infection of OE cells, and the group includes IFN-γ, TNF, IL-1β, OSM, IL-1α, IL-17A, TNFSF-11, IL-1, IL-6, CD40LG, TNFSF-12, CSF-2, IL-18, IL-2, EDN1, CCL5, IL-15, MIF, IL-17F, CXCL2, IL-5, CXCL3, C5, CCL2, CXCL12, IL-7, IL-33, CXCL-8, TNFSF-14, TNFSF-13B, and IL-36A. Our data showed that these 31 cytokines were activated ≥ 2.5-fold in Cm-infected OE-WT cells when compared to the Mock-infected controls. In corroboration with our previously published results [25, 26, 59], many of these cytokines were predicted to be either severely reduced or inhibited in Cm-infected OE-TLR3KO cells relative to Cm-infected OE-WT cells in IPA. It is noteworthy that one particular cytokine (IL-10) was different from the other cytokines listed in Supplementary Table 3 in that it was predicted to be down regulated in the Cm-infected OE-WT cells relative to its expression in the Mock control OE-WT cells, but predicted to be activated in Cm-OE-TLR3KO cells relative to Cm-infected OE-WT cells. We previously reported significant increases in the genital tract secretion of IL-10 of TLR3-deficient mice when compared to wild-type control mice [59]; thus, our findings in the IPA support our in vivo data regarding this pleiotropic regulatory cytokine [60]. We hypothesized that IL-10 plays a role in attenuating the in vivo synthesis of IFN-β during in TLR3-deficient mice and likely promotes the increased Cm-replication that we have observed in the TLR3-deficient mice.
Upstream regulator analyses showed 61 chemical regulators that were either activated or inhibited in Cm-infected OE-WT cells when compared to Mock-infected OE-WT cells, but were differentially regulated in the Cm-infected OE-TLR3KO cells. The chemical categories encompass endogenous non-mammal, endogenous mammal, other, drug, reagent, toxicant, and kinase inhibitors. Further analyses showed that 17 kinase regulators were activated i.e.; CHUK, IKBKB, IKBKG, MAP3K7, RIPK2, MAPKAPK2, JAK2, JAK1, MAP3K14, MAPK8, PRKCD, MAP2K1, PRKCA, SRC, RET, GSK3B, SPHK1, whereas; one kinase (MAPK1) was found to be downregulated in Cm-infected OE-WT cells. Among transcription factors (TFs), 16TFs: NF-κB [complex], REL-A, JUN, STAT4, FOXL2, CEBPB, EGR1, TP63, ECSIT, HMGB1, IRF6, FOXO1, CEBPA, HIF1A, CEBPE, and JUNB) were activated and one TF (ZFP36) was inhibited. Other upstream regulators activated during Cm infection of OE-WT cells included 10 transmembrane receptors (TLR4, TLR3, TLR2, TLR5, CD40, CD14, TNFRSF1A, ICAM1, IL17RA, and TNFRSF1B), one translation regulator (EIF4E), and two transporters (LCN2 and TNNI3).The remaining upstream regulators spanning the 7 categories shown in Supplementary Table 3 were all inhibited in Cm-infected OE-WT cells when compared to Mock-infected OE-WT cells. However, they were also differentially regulated when compared to Cm-infected OE-TLR3KO cells in that they were either inhibited to a lesser degree, or the pathways were activated instead.
Disease pathogenesis and cellular function
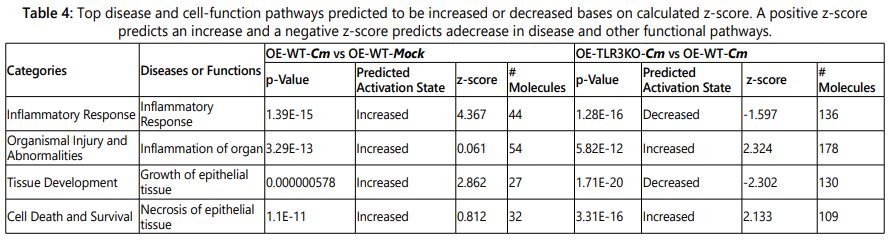
Based on the gene expression input data, IPA predicted the pathways associated with disease pathogenesis and cellular function that are most affected in the cell during Cm infection of OE cells (Table 4). The inflammatory response and growth of epithelial tissue were not surprisingly the most affected pathways that are predicted to be affected when Chlamydia invades epithelial cells lining the female genital tract. As shown in Table 4, these pathways are the most highly up regulated in the Cm-infected OE-WT cells when compared to the Mock-infected OE-WT controls. As indicated, these two pathways are predicted to be significantly diminished or inhibited in the Cm-infected OE-TLR3KO cells when compared to the Cm-infected OE-WT cells. In support of our previously published reports showing that TLR3-deficiency leads to increased genital tract pathology in mice during Cm-infection [59], IPA showed that the pathways associated with organ inflammation and tissue necrosis were predicted to be significantly increased in the Cm-infected OE-TLR3KO cells when compared to Cm-infected OE-WT cells. Collectively, transcriptome and IPA results show role for TLR3 in modulating host cellular responses to Cm infection that extend beyond inflammation and fibrosis, and implicate TLR3 as a major component in the cellular response to Chlamydia infection in genital tract epithelial cells.
Discussion
We previously showed that Cm infection induces IFN-β secretion in OE cells in a mostly TLR3-dependent manner, and that TLR3 deficiency lead to the dysregulation in syntheses of a plethora of other innate inflammatory modulators including IL-6, CXCL10, CXCL16 and CCL5. Although the expression of these cytokines induced by Chlamydia was severely diminished in TLR3 deficient OE cells and mice, replication of Cm during TLR3-deficiency was more robust than in OE-WT cells and mice. These data suggested that TLR3 had a biological impact on the innate immune response to Chlamydia infection; however, the impact of TLR3 signaling on the global cellular response to Chlamydia infection in OE cells remained unanswered. In the present study, we performed transcriptomics on wild-type and TLR3-deficient OE cells and conducted IPA to understand the complexity of genes and pathways in murine OE cells that are affected during Cm infection. In this regard, we are hoping to identify the spectrum of pathways that are either activated or inhibited during Cm infection, in order to ascertain those critical cellular mechanisms that can be exploited for the development of better treatments for Chlamydial disease and therapeutic agents.
Microarray results uncovered a multitude of genes that were either up-or down-regulated ≥ 2.5-fold in Cm infected OE-WT cells when compared to the mock-infected controls, and revealed that many of those genes were differentially regulated during TLR3-deficiency when comparing transcriptome data generated in Cm-infected OE-WT cells to results generated in the Cm-infected OE-TLR3KO cells. Among the most highly up-regulated genes during Cm infection of OE cells, the overwhelming majority were pro-inflammatory cytokines and chemokines (Table 1 and Supplementary Table 3, excel file). Our data showed that many of these pro-inflammatory mediators were either severely attenuated in their expression levels during TLR3 deficiency, or that their expression was down regulated below basal expression levels. Because epithelial cells lining the genital tract serve as immunological sentinels tomicrobial pathogens, and are responsible for initiating the primary phases of the host immune response [19, 21, 23], a comprehensive understanding of the cascade of cellular events that are triggered in OE cell during Cm infection would significantly enhance our knowledge and increase our understanding of the cellular mechanisms induced during Chlamydial pathogenesis.
Cytokines and chemokines are crucial in regulating a variety of molecular and cellular events specifically inflammation, scarring, and fibrosis. We found that CCL5 (RANTES), CXCL2 (MIP2-α), CSF3, CXCL5 (ENA-78), IL-6, CCL4 (MIP-1b), CCL2 (MCP-2) and CCL7 (MCP-3) were among the highest up-regulated cytokine and chemokines. CCL5 showed the highest up-regulation (80.053-fold) in WT-OE cells during Cm infection in comparison to mock-infected WT-OE cells, corroborating results from previous studies [26, 61]. CCL5 is a chemotactic factor that is secreted by a variety of cells including epithelial cells, and functions by recruiting leukocytes to the inflamed sites via binding to its CCR3 receptor. CCL5 also recruits macrophages to the site of inflammation by binding the CCR5 receptor. CCL5 is also a key chemokine in the induction of other Th1 cytokines, and has a functional role in the humoral immune responses against chlamydial pathogens [62, 63]. The C-X-C motif chemokine ligand 2, CXCL2 (MIP2-α) was also shown to be highly induced and was the next highest upregulated chemokine behind CCL5 in the microarray analyses. CXCL2 (along with CCL4) has been demonstrated to be involved in the recruitment of both ymphocytes and neutrophils to the inflamed sites in mouse model of Chlamydia infection [64, 65]. Other significantly upregulated cytokine and chemokine markers were colony stimulating factor 3 (CSF3), IL-6, CXCL5, CCL2 and CCL7. Induction of CSF3 and CXCL5 has been reported to be involved in attraction of neutrophils and other acute inflammatory cells against Chlamydia infection [18, 21, 66-68]. IL-6 is an important mediator of fever and major component of the acute inflammatory response; however, the role IL-6 synthesized during chlamydial genital tract immunopathology in mice is somewhat lesser defined and variable [69, 70]. CCL2 (MCP-1) and CCL7 (MCP-3) were also up-regulated by Cm infection in OE-WT cells, and was shown to play a crucial role in recruiting neutrophils, macrophage/monocytes, and dendritic cells to the site of chlamydial infection by other researchers [18, 68]. Because all of these highly up regulated cytokines and chemokines were severely down-regulated in TLR3-deficient OE cells when compared to WT-OE cells during Cm infection, the microarray results provides more comprehensive evidence that TLR3 deficiency results in a significantly impaired inflammatory immune response to Cm infection in OE cells, and supports our findings that TLR3 alters the pathogenicity of Chlamydia infection in vivo [59]. In addition to the vast array of inflammatory mediators that were affected by Cm infection in OE cells, there were other classes of genes that were significantly up-regulated as part of the cellular response to infection. IPA data showed that Cm infection in OE-WT cells induced genes encoding the glycoprotein laminin subunit gamma-2(LAMC2), follistatin (FST), matrix metalloproteinase (MM10),epiregulin (EREG), autotaxin (ATX or ENPP2)], fibroblast growth factor23 (FGF23), metalloreductase (STEAP4), MAdCAm, and ICAM1,which are known to function in remodeling of extracellular matrix (ECM) proteins [31]. The ECM provides structural and biochemical support of surrounding cells, and although the actual composition of ECM varies between multicellular structures, properties including cell adhesion, cell-to-cell communication, and cell differentiation are common functions of the ECM [71, 72]. The transcriptome data show that Cm infection in OE-WT cells substantially up regulates these ECM genes, and thereby can trigger or disrupt cellular processes that are essential for processes including cellular growth, wound healing, and fibrosis, but are highly suggestive that Chlamydia infection can also have impact on processes such as cell migration, gene expression, and cellular differentiation in macrophages and other hematopoietically-derived cells [73,74]. Our findings support the investigations of others who have implicated several members of this subset of ECM proteins in various disorders associated with genital tract Chlamydia infections such as oviduct fibrosis and scarring, uterus and oviduct distention, and tubal damage caused by ectopic pregnancy [75-80]. Because the transcriptome data show that TLR3-deficiency causes dysregulation in the gene expression levels of most of the ECM proteins identified by IPA, these data provides insight into a possible mechanism that expounds our recent report demonstrating thatTLR3 deficient mice suffer more severe genital tract pathology than wild-type mice during Cm infection [59].
Other genetic factors that were significantly up regulated during Cm infection of OE-WT cells included a subset of host-cell metabolism related genes encoding LCN2, CMPK2, ARG2, SOD2, ERO1-α, CH25H, FABP4, and AK4. Lipocalin-2 (LCN2) is an iron-sequestering multifunctional protein. Iron is an essential nutrient for many intracellular pathogens including Chlamydia. LCN2 binds to bacterial siderophores and thereby limiting the availability of iron for bacteria, inhibiting their growth, and thus protecting host from intracellular pathogens [81-83]. Intracellular pathogens including Chlamydia induce a strong Th1 immune response, which exerts pro-inflammatory effects by stimulating production of free radicals such as reactive oxygen species (ROS) and reactive nitrogen oxide species (RNOS) [84]. L-arginine is a crucial component for both inducible nitric oxide synthase (iNOS) and ARGase 1/2. It has been hypothesized that the enhanced expression of host SOD2 and ARG2 protects the host cells from the damaging ROS and RNOS [85-87], and both genes were found to be upregulated in our analyses. FABP4 (fatty acid binding protein 4) is primarily expressed in adipocytes and macrophages, and has roles in modulating immune responses and in lipid metabolism [88]. Walenna NF. et al demonstrated that C. pneumonia exploits host FABP4 for lipid metabolism in order to obtain ATP and lipids from the host cell, in order to facilitate its robust replication in adipocytes [89]. Not surprisingly, our findings show that TLR3 deficiency either severely attenuates or down regulated the expression of all of these metabolic factors, and presents the hypothesis that dysregulation of these metabolic factors will have a negative impact on the ability of the OE cells to control chlamydial replication. We showed in our previous reports that Cm replication in OE-TLR3KO cells was significantly more robust, and that the chlamydial inclusions were larger and more aberrantly shaped when compared to infection in OE-WT cells [26]. Collectively, our data show that TLR3 signaling does indeed play a role controlling chlamydial replication within the cell, and the data generated in IPA supports the hypothesis that TLR3 mediates this function by regulating the transcription of many of these host cell metabolism related genes.
Other genes that were highly up-regulated in the Cm-infected OE-WT cells but were differentially regulated in the Cm-infected OE-TLR3KO cells included genes that encode proteins that are involved in other areas of innate immunity such as ARL4D, NEURL3, PTX3, SERPINE1 and SERPINB2. These proteins include GTPases, ligases, and protease inhibitors that play important roles in actin remodeling, tissue repair, and ECM degradation [90-98]. In addition to the genes mentioned above, we observed significant increased expression levels of the interferon-responsive genes Gbp5 (a member of IFN-inducible subfamily of GTPases) and Parp12. GBP5 protects host against diverse pathogens [33, 99] and PARP12 localizes to the stress granules under stress condition, mediates cell survival/growth, and induces an anti-viral response by inhibiting protein translation at both viral and cellular protein levels [34-37]. We also saw up-regulated expression of the pro-apoptotic genes Fas and Xaf1 (XIAP associated factor 1) in our analysis, and increased transcription of genes encoding proteins involved in antigen processing including TAP1, LMP7, and LMP2.
We found 8 genes that were induced during Cm infection in OE-WT cells but were not reduced or inhibited in the Cm-infected OE-TLR3KO cells when compared to the Cm-infected WT-OE cells. In contrast to the large majority of genes that were either down regulated or inhibited in TLR3ʼs absence, Isg15, Mx1, Gm4951, Oas1b, Mpeg1, Zfp296 and Klhl2 were all significantly up regulated in Cm-infected TLR3-deficient OE cells when compared to Cm-infected OE-WT cells. Because the absence of TLR3 results in the increased expression of these genes during Chlamydia infection, these results suggest a repressor role for TLR3 in the transcription of these particular genes in OE cells. The repressor function of TLR3 was also observed in the expression of Aldh1a1, Ankrd35, Chchd10, Daam2, Fbln5, Fbxo31, Gcnt1, Lum, Ppm1e, Ptprv and Txnip in Cm-infected OE-WT cells. These genes were either significantly up regulated in the TLR3-deficient OE cells, or their expression levels no longer repressed as they were in OE-WT cells (see Table 2). Although the role of TLR3 as an activator of gene expression has been well described such as in the direct activation of IFN-β in response to dsRNA [100], or in the indirect activation of CCL5 via TLR3-induced IFN-β [101], a mechanism to describe TLR3 signaling as a repressor pathway remains unclear and requires further study.
IPA identified several inflammatory pathways such as acute phase response signaling, dendritic cell maturation, role of PRR in recognition of bacteria and viruses, IL-6 signaling, interferon signaling, TREM1 signaling, VDR/RXR-activation, p38 MAPK signaling, LPS-stimulated MAPK signaling, Th1 signaling, LPS/IL-1 mediated inhibition of RXR function, JAK/STAT signaling, HMGB1 signaling, PI3K/AKT signaling, Tec kinase signaling and TLR signaling pathway to be most crucial in Chlamydia pathogenesis. These pathways were predicted to be significantly activated in Cm-infected OE-WT cells, but were predicted to be either severely attenuated or inhibited in TLR3-deficient OE cells during Cm infection. In contrast, we also found some canonical pathways that were predicted to be inactivated in Cm-infected OE-WT cells such as LXR/RXR activation, PPAR signaling and PPARα/RXRα activation pathway, but were activated in OE cells by Cm during TLR3-deficiency (LXR/RXR activation and PPAR signaling). Not surprisingly, there were several genes affected by Cm infection that are common amongst the various activated and inhibited pathways suggesting that many of these gene products are pleiotropic in their function; however, their impact in their respective pathways were differentially impacted based on the presence or absence of TLR3. These IPA data corroborates the hypothesis that TLR3 functions as a regulator in cellular responses to Chlamydia infection in OE cells, and can function as either an activator or repressor of numerous pathways associated with the inflammatory immune response.
Finally, IPA can be used to identify a cascade of upstream transcriptional regulators from transcriptome data identifying the genes that are activated or repressed during Cm infection of OE cells. In our analyses, we identified a multitude of upstream regulators that were either activated or repressed during Cm infection of OE cells including immunological (cytokines and chemokines), chemical, kinase, transcription factor, transmembrane receptor, translation regulator, and transporter regulators. Among the most highly activated upstream regulators, cytokines were at the top of the list, suggesting Chlamydia was able to modulate the cellular immune responses by altering the expression patterns of some critical immune system molecules including cytokines. Additionally, the main anti-inflammatory-cytokine, IL-10, was shown to be down regulated in Cm-infected OE-WT cells whereas; it was significantly activated in the Cm-infected TLR3-deficient OE cells which supports our recently published in vivo results [59]. IL-10 has been identified as a key player in the establishment and perpetuation of viral persistence, is known to be negatively regulated by IFN-β production, and promotes a suppressive environment that diminishes the antiviral response [102-104]. Because the Cm-induced synthesis of IFN-β is severely diminished in TLR3-deficient OE cells, IPA showing that IL-10 synthesis was significantly up regulated during Cm infection of OE-TLR3KO cells fits the paradigm that its synthesis is regulated by TLR3-dependent IFN-β.
IPA results show that other upstream-activated regulators belonging to classes of kinases, transcription factors, transmembrane receptors, translation regulators, and transporters groups were also impacted by Cm infection in OE cells. As was demonstrated with the immunological regulators, these additional upstream regulators were also negatively regulated in the TLR3-deficient OE cells during Cm infection when compared with Cm-infected OE-WT cells. Further, IPA results show that the Cm infection induced regulators either activated or inhibited cellular pathways in OE-WT cells that were more likely to restrict chlamydial pathogenesis. The IPA data was suggestive that the upstream regulators affected during Cm infection of OE-WT cells were more likely to attenuate chlamydial replication and promote growth of epithelial tissue; whereas, Cm infection in TLR3-deficient OE cells showed activation or inhibition in upstream regulators controlling 3-4 times the number of pathways involved in organ inflammation, fibrosis, and necrosis of epithelial tissue than that in the Cm-infected OE-WT cells. Collectively, the transcriptome and IPA results further validates our previous findings that TLR3 could elicit and regulate host protective cellular responses that limit bacterial proliferation and genital tract pathologies caused by Chlamydia infection [26, 59], and implicate TLR3 as a potential therapeutic target for drug and/ or vaccine development.
Acknowledgements
The authors wish to thank Dr. Santosh K. Maurya of Sanford Burnham Prebys Medical Discovery Institute at Lake Nona, Orlando, Florida, USA for the IPA analysis. This work was supported by National Institutes of Health (NIH) Grant AI104944 and funding from IUSM/CTSI Biomedical Research Grant.
Competing interests
The authors have declared that no competing interests exist.
References
- Gavin L, MacKay AP, Brown K, et al. Sexual and reproductive health of persons aged 10-24 years - United States, 2002-2007. MMWR Surveill Summ. 2009; 58(6): 1-58.
- Paavonen J. Pelvic inflammatory disease. From diagnosis to prevention. Dermatol Clin. 1998; 16(4): 747-56.
- Westrom L, Mardh PA. Current views on the concept of pelvic inflammatory disease. Aust N Z J Obstet Gynaecol. 1984; 24(2): 98-105. doi: 10.1111/j.1479-828X.1984.tb01467.x
- Moller BR, Westrom L, Ahrons S, et al. Chlamydia trachomatis infection of the Fallopian tubes. Histological findings in two patients. Br J Vener Dis. 1979; 55(6): 422-8.
- McCormack WM, Alpert S, McComb DE, et al. Fifteen-month follow-up study of women infected with Chlamydia trachomatis. N Engl J Med. 1979; 300(3): 123-5. doi: 10.1056/NEJM197901183000305
- Ziklo N, Huston WM, Hocking JS, Timms P. Chlamydia trachomatis Genital Tract Infections: When Host Immune Response and the Microbiome Collide. Trends Microbiol. 2016; 24(9): 750-65. doi: 10.1016/j.tim.2016.05.007
- Derbigny WA, Kerr MS, Johnson RM. Pattern recognition molecules activated by Chlamydia muridarum infection of cloned murine oviduct epithelial cell lines. J Immunol. 2005; 175(9): 6065-75. doi: 10.4049/jimmunol.175.9.6065
- Medzhitov R, Janeway CA, Jr. Innate immune recognition and control of adaptive immune responses. Semin Immunol. 1998; 10(5): 351-3.
- Pasare C, Medzhitov R. Toll-dependent control mechanisms of CD4 T cell activation. Immunity. 2004; 21(5): 733-41. doi: 10.1016/j.immuni.2004.10.006
- Hafner LM. Pathogenesis of fallopian tube damage caused by Chlamydia trachomatis infections. Contraception. 2015; 92(2): 108-15. doi: 10.1016/j.contraception.2015.01.004
- Ingalls RR, Rice PA, Qureshi N, Takayama K, Lin JS, Golenbock DT. The inflammatory cytokine response to Chlamydia trachomatis infection is endotoxin mediated. Infect Immun. 1995; 63(8): 3125-30.
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006; 124(4): 783-801. doi: 10.1016/j.cell.2006.02.015
- Barton GM, Medzhitov R. Toll-like receptors and their ligands. Curr Top Microbiol Immunol. 2002; 270: 81-92.
- Aderem A. Role of Toll-like receptors in inflammatory response in macrophages. Crit Care Med. 2001; 29(7 Suppl): S16-8.
- Kopp EB, Medzhitov R. The Toll-receptor family and control of innate immunity. Curr Opin Immunol. 1999; 11(1): 13-8.
- Medzhitov R, Janeway C, Jr. The Toll receptor family and microbial recognition. Trends Microbiol. 2000; 8(10): 452-6.
- Johnson RM. Murine oviduct epithelial cell cytokine responses to Chlamydia muridarum infection include interleukin-12-p70 secretion. Infect Immun. 2004; 72(7): 3951-60. doi: 10.1128/IAI.72.7.3951-3960.2004
- Rasmussen SJ, Eckmann L, Quayle AJ, Shen L, Zhang YX, Anderson DJ, et al. Secretion of proinflammatory cytokines by epithelial cells in response to Chlamydia infection suggests a central role for epithelial cells in chlamydial pathogenesis. J Clin Invest. 1997; 99(1): 77-87. doi: 10.1172/JCI119136
- Commins SP, Borish L, Steinke JW. Immunologic messenger molecules: cytokines, interferons, and chemokines. J Allergy Clin Immunol. 2010; 125(2 Suppl 2): S53-72. doi: 10.1016/j.jaci.2009.07.008
- Darville T, Andrews CW, Sikes JD, Fraley PL, Rank RG. Early local cytokine profiles in strains of mice with different outcomes from chlamydial genital tract infection. Infect Immun. 2001;69(6):3556-61. doi: 10.1128/IAI.69.6.3556-3561.2001
- Johnson RM, Yu H, Strank NO, Karunakaran K, Zhu Y, Brunham RC. B Cell Presentation of Chlamydia Antigen Selects Out Protective CD4gamma13 T Cells: Implications for Genital Tract Tissue-Resident Memory Lymphocyte Clusters. Infect Immun. 2018; 86(2). doi: 10.1128/IAI.00614-17
- Kagnoff MF, Eckmann L. Epithelial cells as sensors for microbial infection. J Clin Invest. 1997; 100(1): 6-10. doi: 10.1172/JCI119522
- Derbigny WA, Hong SC, Kerr MS, Temkit M, Johnson RM. Chlamydia muridarum infection elicits a beta interferon response in murine oviduct epithelial cells dependent on interferon regulatory factor 3 and TRIF. Infect Immun. 2007; 75(3): 1280-90. doi: 10.1128/IAI.01525-06
- Derbigny WA, Johnson RM, Toomey KS, Ofner S, Jayarapu K. The Chlamydia muridarum-induced IFN-beta response is TLR3-dependent in murine oviduct epithelial cells. J Immunol. 2010; 185(11): 6689-97. doi: 10.4049/jimmunol.1001548
- Derbigny WA, Shobe LR, Kamran JC, Toomey KS, Ofner S. Identifying a role for Toll-like receptor 3 in the innate immune response to Chlamydia muridarum infection in murine oviduct epithelial cells. Infect Immun. 2012; 80(1): 254-65. doi: 10.1128/IAI.05549-11
- Schachter J, Caldwell HD. Chlamydiae. Annual review of microbiology. 1980; 34: 285-309. Epub 1980/01/01. doi: 10.1146/annurev.mi.34.100180.001441
- Caldwell HD, Kromhout J, Schachter J. Purification and partial characterization of the major outer membrane protein of Chlamydia trachomatis. Infect Immun. 1981; 31(3): 1161-76.
- Kramer A, Green J, Pollard J, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014; 30(4): 523-30. Epub 2013/12/18. doi: 10.1093/bioinformatics/btt703
- Lu P, Takai K, Weaver VM, Werb Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol. 2011; 3(12). doi: 10.1101/cshperspect.a005058
- Kim BH, Shenoy AR, Kumar P, Bradfield CJ, MacMicking JD. IFN-inducible GTPases in host cell defense. Cell Host Microbe. 2012;12(4):432-44. doi: 10.1016/j.chom.2012.09.007
- Krapp C, Hotter D, Gawanbacht A. et al. Guanylate Binding Protein (GBP) 5 Is an Interferon-Inducible Inhibitor of HIV-1 Infectivity. Cell Host Microbe. 2016; 19(4): 504-14. doi: 10.1016/j.chom.2016.02.019
- Atasheva S, Frolova EI, Frolov I. Interferon-stimulated poly(ADP-Ribose) polymerases are potent inhibitors of cellular translation and virus replication. J Virol. 2014; 88(4): 2116-30. doi: 10.1128/JVI.03443-13
- Catara G, Grimaldi G, Schembri L. et al. PARP1-produced poly-ADP-ribose causes the PARP12 translocation to stress granules and impairment of Golgi complex functions. Sci Rep. 2017; 7(1): 14035. doi: 10.1038/s41598-017-14156-8
- Salazar JC, Duhnam-Ems S, La Vake C. et al. Activation of human monocytes by live Borrelia burgdorferi generates TLR2-dependent and -independent responses which include induction of IFN-beta. PLoS Pathog. 2009; 5(5): e1000444. doi: 10.1371/journal.ppat.1000444
- Welsby I, Hutin D, Gueydan C, Kruys V, Rongvaux A, Leo O. PARP12, an interferon-stimulated gene involved in the control of protein translation and inflammation. J Biol Chem. 2014; 289(38): 26642-57. doi: 10.1074/jbc.M114.589515
- Lad SP, Fukuda EY, Li J, de la Maza LM, Li E. Up-regulation of the JAK/STAT1 signal pathway during Chlamydia trachomatis infection. J Immunol. 2005; 174(11): 7186-93
- Ye N, Ding Y, Wild C, Shen Q, Zhou J. Small molecule inhibitors targeting activator protein 1 (AP-1). J Med Chem. 2014; 57(16): 6930-48. doi: 10.1021/jm5004733
- Delprato A, Al Kadri Y, Perebaskine N, et al. Crucial role of the Rcl1pBms1p interaction for yeast pre-ribosomal RNA processing. Nucleic Acids Res. 2014; 42(15): 10161-72. doi: 10.1093/nar/gku682
- Chiliveru S, Birkelund S, Paludan SR. Induction of interferon-stimulated genes by Chlamydia pneumoniae in fibroblasts is mediated by intracellular nucleotide-sensing receptors. PLoS One. 2010; 5(4): e10005. doi: 10.1371/journal.pone.0010005
- Gruenheid S, Gros P. Forward genetic dissection of innate response to infection in inbred mouse strains: selected success stories. Clin Exp Immunol. 2010; 162(3): 393-401. doi: 10.1111/j.1365-2249.2010.04249.x
- McCormack RM, Szymanski EP, Hsu AP, et al. MPEG1/perforin-2 mutations in human pulmonary nontuberculous mycobacterial infections. JCI Insight. 2017; 2(8). doi: 10.1172/jci.insight.89635
- Radoshevich L, Impens F, Ribet D, et al. ISG15 counteracts Listeria monocytogenes infection. Elife. 2015; 4. doi: 10.7554/eLife.06848
- Samuel MA, Diamond MS. Pathogenesis of West Nile Virus infection: a balance between virulence, innate and adaptive immunity, and viral evasion. J Virol. 2006; 80(19): 9349-60. doi: 10.1128/JVI.01122 06
- Chen HY, Zhu BH, Zhang CH, et al. High CpG island methylator phenotype is associated with lymph node metastasis and prognosis in gastric cancer. Cancer Sci. 2012; 103(1): 73-9. doi: 10.1111/j.1349-7006.2011.02129.x
- Chen MB, Liu YY, Cheng LB, Lu JW, Zeng P, Lu PH. AMPKalpha phosphatase Ppm1E upregulation in human gastric cancer is required for cell proliferation. Oncotarget. 2017; 8(19): 31288-96. doi: 10.18632/oncotarget.16126
- Galamb O, Kalmar A, Peterfia B, et al. Aberrant DNA methylation of WNT pathway genes in the development and progression of CIMP-negative colorectal cancer. Epigenetics. 2016; 11(8): 588-602. doi: 10.1080/15592294.2016.1190894
- Hatakeyama S, Yoneyama T, Tobisawa Y, Ohyama C. Recent progress and perspectives on prostate cancer biomarkers. Int J Clin Oncol. 2017; 22(2): 214-21. doi: 10.1007/s10147-016-1049-y
- Huang HL, Jiang Y, Wang YH, et al. FBXO31 promotes cell proliferation, metastasis and invasion in lung cancer. Am J Cancer Res. 2015; 5(5): 1814-22.
- Keymoosi H, Gheytanchi E, Asgari M, Shariftabrizi A, Madjd Z. ALDH1 in combination with CD44 as putative cancer stem cell markers are correlated with poor prognosis in urothelial carcinoma of the urinary bladder. Asian Pac J Cancer Prev. 2014; 15(5): 2013-20.
- Lavrov AV, Chelysheva EY, Smirnikhina SA, et al. Frequent variations in cancer-related genes may play prognostic role in treatment of patients with chronic myeloid leukemia. BMC Genet. 2016; 17 Suppl 1:14. doi: 10.1186/s12863-015-0308-7
- Leinung M, Ernst B, Doring C, et al. Expression of ALDH1A1 and CD44 in primary head and neck squamous cell carcinoma and their value for carcinogenesis, tumor progression and cancer stem cell identification. Oncol Lett. 2015; 10(4): 2289-94. doi: 10.3892/ol.2015.3542.
- Topalovski M, Hagopian M, Wang M, Brekken RA. Hypoxia and Transforming Growth Factor beta Cooperate to Induce Fibulin-5 Expression in Pancreatic Cancer. J Biol Chem. 2016; 291(42): 22244-52. doi: 10.1074/jbc.M116.730945
- Doumont G, Martoriati A, Marine JC. PTPRV is a key mediator of p53-induced cell cycle exit. Cell Cycle. 2005; 4(12): 1703-5. doi: 10.4161/cc.4.12.2207
- Park YJ, Yoon SJ, Suh HW, et al. TXNIP deficiency exacerbates endotoxic shock via the induction of excessive nitric oxide synthesis. PLoS Pathog. 2013; 9(10): e1003646. doi: 10.1371/journal.ppat.1003646
- Shao H, Lee S, Gae-Scott S, et al. Extracellular matrix lumican promotes bacterial phagocytosis, and Lum-/- mice show increased Pseudomonas aeruginosa lung infection severity. J Biol Chem. 2012; 287(43): 35860-72. doi: 10.1074/jbc.M112.380550
- Sullivan AB, Tam KP, Metruccio MM, Evans DJ, Fleiszig SM. The importance of the Pseudomonas aeruginosa type III secretion system in epithelium traversal depends upon conditions of host susceptibility. Infect Immun. 2015; 83(4): 1629-40. doi: 10.1128/IAI.02329-14
- Carrasco SE, Hu S, Imai DM, Kumar R, Sandusky GE, Yang XF, et al. Toll-like receptor 3 (TLR3) promotes the resolution of Chlamydia muridarum genital tract infection in congenic C57BL/6N mice. PLoS One. 2018; 13(4): e0195165. doi: 10.1371/journal.pone.0195165
- Mannino MH, Zhu Z, Xiao H, Bai Q, Wakefield MR, Fang Y. The paradoxical role of IL-10 in immunity and cancer. Cancer Lett. 2015; 367(2): 103-7. Epub 2015/07/19. doi: 10.1016/j.canlet.2015.07.009
- Porcella SF, Carlson JH, Sturdevant DE, et al. Transcriptional profiling of human epithelial cells infected with plasmid-bearing and plasmiddeficient Chlamydia trachomatis. Infect Immun. 2015; 83(2): 534-43. doi: 10.1128/IAI.02764-14
- Appay V, Rowland-Jones SL. RANTES: a versatile and controversial chemokine. Trends Immunol. 2001; 22(2): 83-7.
- Sakthivel SK, Singh UP, Singh S, Taub DD, Igietseme JU, Lillard JW. CCL5 regulation of mucosal chlamydial immunity and infection. BMC Microbiol. 2008; 8: 136. doi: 10.1186/1471-2180-8-136
- Moore-Connors JM, Fraser R, Halperin SA, Wang J. CD4(+)CD25(+)Foxp3(+) regulatory T cells promote Th17 responses and genital tract inflammation upon intracellular Chlamydia muridarum infection. J Immunol. 2013; 191(6): 3430-9. doi: 10.4049/jimmunol.1301136
- Ohtsuka Y, Lee J, Stamm DS, Sanderson IR. MIP-2 secreted by epithelial cells increases neutrophil and lymphocyte recruitment in the mouse intestine. Gut. 2001; 49(4): 526-33.
- Lee HY, Schripsema JH, Sigar IM, et al. A role for CXC chemokine receptor-2 in the pathogenesis of urogenital Chlamydia muridarum infection in mice. FEMS Immunol Med Microbiol. 2010; 60(1): 49-56. doi: 10.1111/j.1574-695X.2010.00715.x
- Miyairi I, Tatireddigari VR, Mahdi OS, et al. The p47 GTPases Iigp2 and Irgb10 regulate innate immunity and inflammation to murine Chlamydia psittaci infection. J Immunol. 2007; 179(3): 1814-24.
- Rank RG, Lacy HM, Goodwin A, et al. Host chemokine and cytokine response in the endocervix within the first developmental cycle of Chlamydia muridarum. Infect Immun. 2010; 78(1): 536-44. doi: 10.1128/IAI.00772-09
- Cunningham K, Stansfield SH, Patel P, et al. The IL-6 response to Chlamydia from primary reproductive epithelial cells is highly variable and may be involved in differential susceptibility to the immunopathological consequences of chlamydial infection. BMC immunology. 2013; 14:50. doi: 10.1186/1471-2172-14-50
- Sun X, Tian Q, Wang L, Xue M, Zhong G. IL-6-mediated signaling pathways limit Chlamydia muridarum infection and exacerbate its pathogenicity in the mouse genital tract. Microbes and infection. 2017; 19(11): 536-45. doi: 10.1016/j.micinf.2017.08.007
- Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nature Reviews Molecular Cell Biology. 2014; 15:786. doi: 10.1038/nrm3904
- Theocharis AD, Skandalis SS, Gialeli C, Karamanos NK. Extracellular matrix structure. Advanced Drug Delivery Reviews. 2016; 97: 4-27. doi: https://doi.org/10.1016/j.addr.2015.11.001
- Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix Elasticity Directs Stem Cell Lineage Specification. Cell. 2006; 126(4): 677-89. doi: 10.1016/j.cell.2006.06.044
- Wang JHC, Thampatty BP, Lin JS, Im HJ. Mechanoregulation of gene expression in fibroblasts. Gene. 2007; 391(1): 1-15. doi: https://doi.org/10.1016/j.gene.2007.01.014
- Davila SJ, Olive AJ, Starnbach MN. Integrin alpha4beta1 is necessary for CD4+ T cell-mediated protection against genital Chlamydia trachomatis infection. J Immunol. 2014; 192(9): 4284-93. doi: 10.4049/jimmunol.1303238
- Igietseme JU, Portis JL, Perry LL. Inflammation and clearance of Chlamydia trachomatis in enteric and nonenteric mucosae. Infect Immun. 2001; 69(3): 1832-40. doi: 10.1128/IAI.69.3.1832-1840.2001
- Kelly KA, Chan AM, Butch A, Darville T. Two different homing pathways involving integrin beta7 and E-selectin significantly influence trafficking of CD4 cells to the genital tract following Chlamydia muridarum infection. Am J Reprod Immunol. 2009; 61(6): 438-45. doi: 10.1111/j.1600-0897.2009.00704.x
- Refaat BA, Bahathiq AO, Sockanathan S, Stewart RL, Wells M, Ledger WL. Production and localization of activins and activin type IIA and IIB receptors by the human endosalpinx. Reproduction. 2004; 128(2): 249-55. doi: 10.1530/rep.1.00156
- Refaat B, Al-Azemi M, Geary I, Eley A, Ledger W. Role of activins and inducible nitric oxide in the pathogenesis of ectopic pregnancy in patients with or without Chlamydia trachomatis infection. Clin Vaccine Immunol. 2009; 16(10): 1493-503. doi: 10.1128/CVI.00221-09
- Jansen AFM, Schoffelen T, Textoris J, et al. Involvement of matrix metalloproteinases in chronic Q fever. Clin Microbiol Infect. 2017; 23(7): 487 e7- e13. doi: 10.1016/j.cmi.2017.01.022
- Bellmann-Weiler R, Schroll A, Engl S, et al. Neutrophil gelatinaseassociated lipocalin and interleukin-10 regulate intramacrophage Chlamydia pneumoniae replication by modulating intracellular iron homeostasis. Immunobiology. 2013; 218(7): 969-78. doi: 10.1016/j.imbio.2012.11.004
- Hop HT, Arayan LT, Huy TXN, et al. Lipocalin 2 (Lcn2) interferes with iron uptake by Brucella abortus and dampens immunoregulation during infection of RAW 264.7 macrophages. Cell Microbiol. 2018; 20(3). doi: 10.1111/cmi.12813
- Zhao H, Konishi A, Fujita Y, et al. Lipocalin 2 bolsters innate and adaptive immune responses to blood-stage malaria infection by reinforcing host iron metabolism. Cell Host Microbe. 2012; 12(5): 705-16. doi: 10.1016/j.chom.2012.10.010
- Swindle EJ, Metcalfe DD. The role of reactive oxygen species and nitric oxide in mast cell-dependent inflammatory processes. Immunol Rev. 2007; 217: 186-205. doi: 10.1111/j.1600-065X.2007.00513.x
- Eisenreich W, Heesemann J, Rudel T, Goebel W. Metabolic host responses to infection by intracellular bacterial pathogens. Front Cell Infect Microbiol. 2013; 3: 24. doi: 10.3389/fcimb.2013.00024
- Prusty BK, Bohme L, Bergmann B, et al. Imbalanced oxidative stress causes chlamydial persistence during non-productive human herpes virus coinfection. PLoS One. 2012; 7(10): e47427. doi: 10.1371/journal. pone.0047427
- Wang C, van Ginkel FW, Kim T, et al. Temporal delay of peak T-cell immunity determines Chlamydia pneumoniae pulmonary disease in mice. Infect Immun. 2008; 76(11): 4913-23. doi: 10.1128/IAI.00569-08
- Storch J, Thumser AE. Tissue-specific functions in the fatty acid-binding protein family. J Biol Chem. 2010; 285(43): 32679-83. doi: 10.1074/jbc.R110.135210
- Walenna NF, Kurihara Y, Chou B, Ishii K, Soejima T, Itoh R, et al. Chlamydia pneumoniae exploits adipocyte lipid chaperone FABP4 to facilitate fat mobilization and intracellular growth in murine adipocytes. Biochem Biophys Res Commun. 2018; 495(1): 353-9. doi: 10.1016/j.bbrc.2017.11.005
- Li CC, Wu TS, Huang CF, et al. GTP-binding-defective ARL4D alters mitochondrial morphology and membrane potential. PLoS One. 2012; 7(8): e43552. doi: 10.1371/journal.pone.0043552
- Hu Y, Nguyen TT, Bui KC, Demello DE, Smith JB. A novel inflammationinduced ubiquitin E3 ligase in alveolar type II cells. Biochem Biophys Res Commun. 2005; 333(1): 253-63. doi: 10.1016/j.bbrc.2005.05.102
- Jaillon S, Moalli F, Ragnarsdottir B, et al. The humoral pattern recognition molecule PTX3 is a key component of innate immunity against urinary tract infection. Immunity. 2014; 40(4): 621-32. doi: 10.1016/j.immuni.2014.02.015
- Magrini E, Mantovani A, Garlanda C. The Dual Complexity of PTX3 in Health and Disease: A Balancing Act?. Trends Mol Med. 2016; 22(6): 497-510. doi: 10.1016/j.molmed.2016.04.007
- Harslund J, Frees D, Leifsson PS, Offenberg H, Romer MU, Brunner N, et al. The role of Serpine-1 and Tissue inhibitor of metalloproteinase type-1 in early host responses to Staphylococcus aureus intracutaneous infection of mice. Pathog Dis. 2013; 68(3): 96-104. doi: 10.1111/2049-632X.12055
- Major LD, Partridge TS, Gardner J, et al. Induction of SerpinB2 and Th1/Th2 modulation by SerpinB2 during lentiviral infections in vivo. PLoS One. 2013; 8(2): e57343. doi: 10.1371/journal.pone.0057343
- Renckens R, Roelofs JJ, Bonta PI, et al. Plasminogen activator inhibitor type 1 is protective during severe Gram-negative pneumonia. Blood. 2007; 109(4): 1593-601. doi: 10.1182/blood-2006-05-025197
- Schroder WA, Le TT, Major L, et al. A physiological function of inflammation-associated SerpinB2 is regulation of adaptive immunity. J Immunol. 2010; 184(5): 2663-70. doi: 10.4049/jimmunol.0902187
- Shea-Donohue T, Zhao A, Antalis TM. SerpinB2 mediated regulation of macrophage function during enteric infection. Gut Microbes. 2014; 5(2): 254-8. doi: 10.4161/gmic.28093
- Pilla-Moffett D, Barber MF, Taylor GA, Coers J. Interferon-Inducible GTPases in Host Resistance, Inflammation and Disease. J Mol Biol. 2016; 428(17): 3495-513. doi: 10.1016/j.jmb.2016.04.032
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of doublestranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001; 413(6857): 732-8.
- Nakano M, Fujii T, Hashimoto M, Yukawa N, Yoshifuji H, Ohmura K, et al. Type I interferon induces CX3CL1 (fractalkine) and CCL5 (RANTES) production in human pulmonary vascular endothelial cells. Clin Exp Immunol. 2012; 170(1): 94-100. Epub 2012/09/05. doi: 10.1111/j.1365-2249.2012.04638.x
- Taylor DR, Shi ST, Romano PR, Barber GN, Lai MM. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science. 1999; 285(5424): 107-10.
- Wilson EB, Brooks DG. The role of IL-10 in regulating immunity to persistent viral infections. Curr Top Microbiol Immunol. 2011; 350: 39-65.
- Zuniga EI, Liou LY, Mack L, Mendoza M, Oldstone MB. Persistent virus infection inhibits type I interferon production by plasmacytoid dendritic cells to facilitate opportunistic infections. Cell Host Microbe. 2008; 4(4): 374-86.
