

Review Article
A “Not-So-Silent” Silent Carrier of Alpha-Thalassemia Presenting with Significant Microcytosis, Anemia, and Erythrocytosis: A Case Study and Literature Review
Department of Pathology, University of Texas Medical Branch, Galveston,+87 TX, USA
*Corresponding author: Jianli Dong, Department of Pathology, University of Texas Medical Branch, 301 University Boulevard, Galveston, TX, USA, E-mail: jidong@utmb.edu
Received: November 2, 2020 Accepted: December 14, 2020 Published: December 18, 2020
Citation: Rapp A, Huang G, Dong J. A “Not-So-Silent” Silent Carrier of AlphaThalassemia Presenting with Significant Microcytosis, Anemia, and Erythrocytosis: A Case Study and Literature Review. Int J Hematol Blood Res. 2020; 1(1): 16-20. doi: 10.18689/ijhbr-1000104
Copyright: © 2020 The Author(s). This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Alpha-thalassemia is a hereditary microcytic, hypochromic anemia characterized by a decrease in the amount of α-globin chains resulting from mutations in one or more of the four α-globin genes. The mode of inheritance is autosomal recessive, with carrier states divided into silent alpha-thalassemia (one affected gene) and alpha-thalassemia trait (two affected genes). It has been established that patients with the silent carrier genotype are asymptomatic with either normal hematologic parameters or occasional mild microcytosis/hypochromia. Herein, we describe a 15-year-old, Hispanic male with an unexplained microcytic anemia with accompanying erythrocytosis and normal iron studies, phenotypically consistent with alpha-thalassemia trait. Genetic testing using polymerase chain reaction (gap-PCR) and multiplex ligation-dependent probe amplification (MLPA) detected a heterozygous -α3.7 deletion, consistent with a diagnosis of alpha-thalassemia silent carrier. This silent carrier with a “non-silent” phenotype suggests the existence of other genetic and/or environmental factors that can modify the phenotypic expression of alpha-thalassemia.
Keywords: Alpha-thalassemia; HBA1; HBA2; Heterozygous carrier; Haploinsufficiency.
Introduction
Alpha-thalassemia is a hereditary, autosomal recessive disorder of hemoglobin (Hb) in which there is decreased production of the α-globin chains of adult and fetal Hb due to alterations in one or more of the four α-globin genes (2 copies of HBA1 and 2 copies of HBA2). The structure of hemoglobin is that of a tetrameric protein comprised of two α-like and two β-like chains that comprise the various types of hemoglobin [1,2]. Normal adult hemoglobin variants include hemoglobin A (HbA), hemoglobin A2 (HbA2), and hemoglobin F (HbF), with HbA consisting of about 97% of total Hb levels. All of these variants consist of two α-globin chains and either two β-globin chains for HbA, two δ-globin chains for HbA2, or two γ-globin chains for HbF. Therefore, as the α-globin chains are common to all these normal variants, any disturbances in α-globin synthesis can detrimentally impact hemoglobin synthesis from fetal through adult life.
Under normal conditions, there are two α-globin genes (HBA1 and HBA2) on each copy of chromosome 16 (16p13.3), with a total of four α-globin genes responsible for α-globin synthesis [1,3]. Deficient expression of these genes creates an unbalanced ratio of α-globin to β-globin. The resulting excess β-globin chains in the adult and excess γ-globin chains in the fetus form insoluble tetramers, called HbH (β4) and Hb Bartʼs (γ4), respectively. These tetramers precipitate in the red blood cell (RBC), leading to hemolysis, ineffective erythropoiesis, and variable degrees of microcytic, hypochromic anemia and extramedullary hematopoiesis.
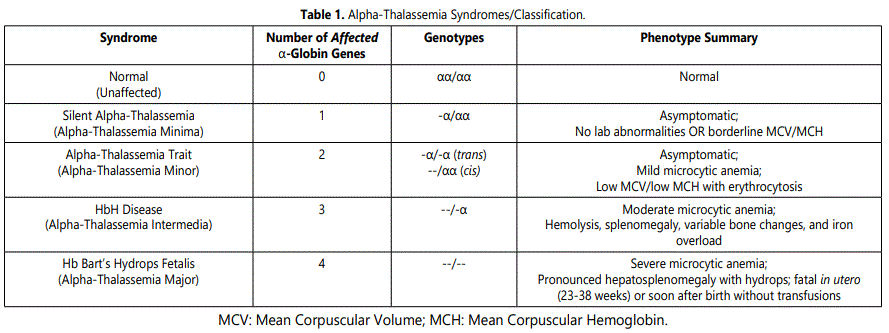
Alpha-thalassemia has a very broad and diverse range of clinical symptoms [1,2] reflective of the number of affected α-globin genes (Table 1). HbH disease and Hb Bartʼs hydrops fetalis syndrome are the two major clinical alpha-thalassemia syndromes, resulting from mutations involving three or four α-globin genes, respectively. HbH disease is characterized by moderate anemia, hemolysis with Heinz bodies, splenomegaly, variable bone changes, and iron overload. Infants with Hb Bartʼs hydrops fetalis syndrome make very little or no α-globin chains and have a very severe anemia which is lethal in the fetal or early neonatal period without transfusions [1].
Additionally, there are two alpha-thalassemia carrier states. In alpha-thalassemia trait, two α-globin genes are defective while two functional genes remain, resulting in a mild microcytic, hypochromic anemia with erythrocytosis that is generally asymptomatic [1,2,4]. In silent alpha-thalassemia, only one α-globin gene is defective. Patients with silent alphathalassemia are said to be silent carriers due to their asymptomatic presentation and lack of significant abnormalities in hematologic parameters, other than possible mild microcytosis and/or borderline hypochromia [1,2,4]. However, recent studies have shown some silent carriers to have clinically significant microcytosis, hypochromia, and erythrocytosis similar to alpha-thalassemia trait [5-7]. Here we present a case study of a 15-year old, Hispanic male with a heterozygous -α3.7 deletion, consistent with silent carrier status, but with marked microcytosis, hypochromia, and erythrocytosis analogous to the alpha-thalassemia trait phenotype. The study was approved by the UTMBʼs Institutional Review Board (IRB #02-089).
Clinical History
One month prior to presentation, a 15-year old, Hispanic male with a history of recurrent epistaxis was discovered to have microcytic anemia after routine laboratory studies revealed a low Hb of 11.2 g/dL and a low mean corpuscular volume (MCV) of 65 fL. Iron studies at that time were within normal limits: serum iron, 116 μg/dL; serum ferritin (SF), 92 ng/mL; total iron-binding capacity (TIBC), 376 μg/dL; and transferrin saturation (TS), 31%. Additionally, all coagulation parameters including measures of von Willebrand factor (VWF) and factor VIII (FVIII) were within normal limits. Due to the patientʼs microcytic anemia in the context of normal iron studies, the patientʼs pediatrician referred him to pediatric hematology at our institution to evaluate for a suspected hemoglobinopathy, such as thalassemia trait.
Upon presentation to the pediatric hematology clinic, the patient reported a history of epistaxis since early childhood with a frequency of one to two episodes per week and duration of approximately five minutes per episode. He stated that these episodes usually were provoked by heat and/or showering and that his left nostril was predominantly affected. Other than epistaxis, he had no other abnormal bleeding tendencies, such as easy bruising or melena/hematochezia, nor symptoms of anemia, such as excessive fatigue, chest pain, or shortness of breath. The only member of his family with anemia was his 13-year-old sister, who at the time was being treated with oral iron therapy. The patient did not have any known family history of bleeding disorders or hemoglobinopathies.
On examination, the patient appeared to be a healthy adolescent male in no acute distress who was alert and oriented. His weight was 83 kg, placing him at the 96th percentile for his age and height. Vital signs were within acceptable parameters. Nasal examination revealed slight septal deviation in the left nostril. Otherwise, the rest of his physical examination yielded no abnormalities.
A complete blood count (CBC) done by our laboratory confirmed the presence of a microcytic anemia: Hb, 11.3 g/dL (13.0-16.0 g/dL); MCV of 66.2 fL (78.0-95.0 fL). Additionally, the CBC revealed a significant erythrocytosis of 6.01 × 106 cells/μL (4.50-5.30 × 106/μL), as well as a markedly low mean corpuscular hemoglobin (MCH) of 18.8 pg (26.0-32.0 pg), and a low mean corpuscular hemoglobin concentration (MCHC) of 28.4 g/dL (32.0-36.0 g/dL). Red cell distribution widthstandard deviation (RDW-SD) also was low at 37.0 fL (38.5-49.0), while the red cell distribution width-coefficient of variation (RDW-CV) was elevated at 16.1% (RDW-CV) (Table 2). Peripheral smear review by our hematopathologist visually confirmed this microcytic, hypochromic anemia, additionally noting the presence of microcytes, ovalocytes, and a few target cells. Hemoglobin electrophoresis by capillary electrophoresis (CE) and high-performance liquid chromatography (HPLC) demonstrated a normal pattern of HbA and HbA2, the lack of elevated HbA2 ruling out betathalassemia trait from the differential diagnosis. Given that the patientʼs iron studies were normal, iron deficiency as the underlying etiology of his microcytosis was excluded. Therefore, alpha-thalassemia trait was suspected.
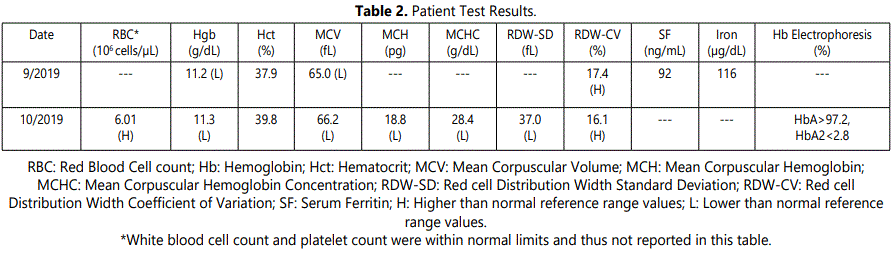
Molecular studies were performed to investigate for suspected alpha-thalassemia. We tested DNA extracted from the patientʼs peripheral blood for the seven most common α-globin deletions using single-tube multiplex polymerase chain reaction (GAP-PCR) methods [8,9]. Additionally, we tested him for additional deletions/duplications using the SALSA multiplex ligation dependent probe amplification (MLPA) probemix P140 HBA assay (MRC Holland, Amsterdam, NL). Both gap-PCR and MLPA revealed the patient to have one copy of the -α3.7 deletion, consistent with a diagnosis of silent alpha-thalassemia.
Laboratory Role in Diagnosis
The diagnosis of alpha-thalassemia primarily relies on a combination of laboratory data, including CBC and iron studies, hemoglobin electrophoresis, and genetic testing. As most affected individuals have some degree of anemia, reduced MCH, reduced MCV, and either erythrocytosis or relative erythrocytosis (in which the RBC count is disproportionally normal in relation to the degree of anemia), alpha-thalassemia is most frequently suspected after a routine CBC demonstrates a microcytic, hypochromic anemia with erythrocytosis in the context of iron sufficiency [1,2]. Combined use of HPLC and capillary electrophoresis is useful to detect abnormal hemoglobin fractions, such as HbH and Hb Bartʼs, seen in the major clinical alpha-thalassemia syndromes. It can also detect other Hb variants to assess for combined hemoglobinopathies, such as combined alphathalassemia sickle-cell traits. Additionally, patients with alpha-thalassemia have normal or slightly reduced HbA2 levels [1], which can be useful in excluding a concomitant beta-thalassemia trait, in which the HbA2 levels are elevated above 3.5% [10]. However, while hemoglobin electrophoresis can make the distinction between these two thalassemias, it cannot differentiate the various alpha-thalassemia syndromes, nor can it definitively diagnosis alpha-thalassemia. Therefore, molecular analysis of the α-globin gene cluster is necessary to confirm the diagnosis, especially since alpha-thalassemia carriers show normal levels of HbA, HbA2, and HbF [1,4].
The α-globin gene family resides on chromosome 16p13.3 [2,3]. The α-globin gene cluster in its entirety contains the following genes in order from 5ʼ to 3ʼ: ζ2-ψζ1-ψα2-ψα1-α2-α1-θ1, where ψζ1, ψα2, and ψα1 are pseudogenes and θ1 has undetermined function [2-4,11]. About 90% of alpha-thalassemia cases are caused by deletions of one (-α) or both (--) α-globin genes from a haploid chromosome 16 [1,4], referred to as α+ and α0 thalassemia deletions, respectively. The most common deletions are the -α3.7 and the -α4.2 single α-globin gene deletions. Common double α-globin gene deletions include --SEA, --FIL, --THAI common in those of Southeast Asian descent and the --MED and the -(α)20.5 most frequently seen in those of Mediterranean descent [1,2]. The remaining small minority of cases are caused by point mutations, usually within the HBA2 gene [12], as well as deletions removing the upstream regulatory elements of the α-globin cluster [1,2].
Polymerase chain reaction (PCR) is the most common method utilized to confirm the diagnosis of deletional α-thalassemia [4], with single-tube multiplex polymerase chain reaction (gap-PCR) developed to detect the seven most common alpha-thalassemia deletions. Since most alpha-thalassemias are deletional, this provides a very quick way to diagnose α+ and α0 thalassemia deletions [11]. Additionally, multiplex ligation dependent probe amplification (MLPA) has become widely used [4]. Since gap-PCR cannot be used to diagnose rare α+ and α0 thalassemia mutations as their breakpoint sequences have not been determined, MLPA can be used to detect both known and unknown deletions. MLPA can also detect gene rearrangements leading to duplication of the α-globin genes in the form of triple and quadruple α-globin gene alleles [13]. Thus, MLPA is a valuable adjunct method to gap-PCR when investigating both known and unknown deletions causing alpha-thalassemia [1].
Patient Follow-up
As his prior coagulation studies were within normal limits and thus inconsistent with coagulopathy, his epistaxis was attributable to either a bone spur or trauma from nose picking, unrelated to his hematologic findings. The patient was advised to return to pediatric hematology in one month and referred to pediatric otolaryngology for his epistaxis and deviated nasal septum. Unfortunately, the patient was lost to subsequent follow-up.
Discussion
Alpha-thalassemia is the most common hemoglobinopathy worldwide [14] and one of the most common human monogenic diseases [1,3], affecting more than 300,000 people born worldwide per year [2]. Of all globin disorders, alphathalassemia has the widest distribution [1]. It occurs at very high frequencies in subtropical and tropical regions, with some areas reaching carrier frequencies as high as 80-90%, likely due to the carrier state providing a selective advantage in malaria-endemic areas [1]. However, as a result of massive global migration patterns over the past several decades, alpha-thalassemiaʼs prevalence in the rest of the world, including Northern Europe and North America, has significantly risen [1,4], thereby becoming increasingly included on the differential diagnosis of microcytic anemia after iron deficiency has been excluded.
Clinical phenotype severity can be predicted based on the number of mutated α-globin genes, with severity directly correlating with the number of genes affected. Along these lines, the degree of microcytosis and hypochromia (as well as other hematologic parameters) can in theory be predicted by the number of affected α-globin genes and vice versa. Investigating the ability of hematologic parameters to predict the underlying genotype, a study in 2017 by Velasco-Rodríguez et al. involving 129 cases of deletional alphathalassemia demonstrated a good correlation between the number of deleted α-globin genes and MCV (r=-0.672, p<0.001), MCH (r=-0.788, p<0.001), and RDW (r=0.633, p<0.001). Notably, they also found that MCH<21.90 pg and/or MCV<70.80 fL was strongly indicative of at least one α0 allele [4], thereby implying a genotype consistent with at least alpha-thalassemia trait. For silent carriers ages 3 to 16 years, MCV was expected to be 75.6 ± 3.82 fL and MCH 24.2 ± 1.07 pg [4]. While distinct cut-off parameters for the various alpha-thalassemia types have yet to be established, it is generally accepted that hematologic parameter abnormalities tend to be more pronounced in alpha-thalassemia trait than in silent alpha-thalassemia [1].
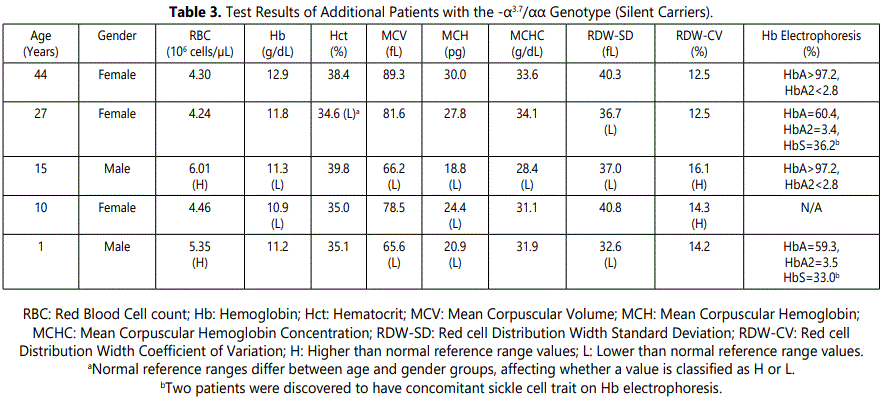
However, it has become increasingly recognized that some alpha-thalassemia silent carriers may have hematologic parameters similar to alpha-thalassemia trait [5,6]. In a recent study by Gilad et al. involving 192 children found to be heterozygous for the -α3.7 deletion, it was found that these silent carriers had significantly lower Hb and MCV than controls and significantly higher RBC counts [7], similar to that found in our patient. Furthermore, the greatest reduction in Hb was seen in adolescent males [7]. We observed similar findings at our institution. In the five, non-iron deficient patients diagnosed with a single -α3.7 deletion, this patient had the greatest number and degree of hematologic abnormalities followed by the infant and 10 year-old child, while two adult patients had no to minimal red blood cell abnormalities (Table 3).
To our knowledge, no previously established literature exists explaining why some heterozygous -α3.7 carriers (silent alpha-thalassemia) show hematologic patterns similar to homozygous -α3.7 carriers (alpha-thalassemia trait). This prompts a closer examination of the molecular mechanisms of alpha-thalassemia and the limitations of diagnostic methods.
As previously stated, alpha-thalassemia is more frequently caused by deletions than non-deletional mutations [1]. However, when they occur, heterozygous non-deletional α+ thalassemia mutations typically result in a more severe reduction in α-globin chain synthesis and a more severe clinical phenotype than heterozygous α+ thalassemia deletions [1,15], likely due to these mutations involving genomic regions that regulate normal expression of the α-globin genes [15]. This raises the possibility that this patient – as well as other silent carriers of alpha-thalassemia with pronounced hematologic abnormalities – have an additional, undetected non-deletional mutation. In the most commonly used diagnostic laboratory methods, gap-PCR only detects the seven most common alpha-thalassemia deletions [1,4,13], so it cannot detect these non-deletional mutations. Furthermore, while MLPA can detect unknown deletions and gene rearrangements leading to duplication of the α-globin genes [1,13], the SALSA MLPA probemix P140 HBA assay used in our laboratory cannot detect point mutations except for the Hb Constant Spring mutation. While the Hb Constant Spring mutation was not detected in this patient, it has been shown that co-inheritance of Hb Constant Spring and a single -α3.7 deletion results in a phenotype similar to at least alphathalassemia trait [16]. Therefore, it is possible that coinheritance of other undetected non-deletional mutations with a single -α3.7 deletion may also lead to an alphathalassemia trait-like phenotype. Thus, this patient – as well as other heterozygous -α3.7 deletion carriers at institutions using similar methodology – might in theory have an additional point mutation undetectable by the current laboratory methods.
Additionally, some autosomal recessive disorders in rare instances have shown heterozygotes to unexpectedly have a clinical phenotype similar to affected homozygotes. An example of this was demonstrated in a case report of a heterozygous C282Y HFE carrier with a family history of hemochromatosis presenting with laboratory evidence of iron overload [17]. While typically C282Y carriers with iron overload are assumed to have other genetic or environmental influences since hemochromatosis is an autosomal recessive disorder, the presented patient did not have any associated additional risk factors for iron overload. Thus the authors concluded that haploinsufficiency of the HFE gene may lead to iron overload in some heterozygous HFE carriers. While to the best of our knowledge isolated alpha-thalassemia syndromes have not been attributed to haploinsufficiency, alpha-thalassemia retardation associated with chromosome 16 syndrome (ATR-16 syndrome) has been associated with haploinsufficiency [18]. This syndrome is characterized by dysmorphic features, mental retardation, and an accompanying alpha-thalassemia, with heterozygous deletions in chromosome 16p that involves HBA1, HBA2, and other genes [18]. However, it is currently uncertain if haploinsufficiency alone or in part can lead to phenotypically expressive silent alpha-thalassemia.
While at this time we cannot definitively explain selectively increased phenotypic expression in some silent alphathalassemia carriers, this case emphasizes the continuously emerging complexities associated with the refinement of the molecular etiologies and diagnosis of alpha-thalassemia. Emerging technologies including next generation sequencing [13] may provide further insight into the intricate molecular anomalies of alpha-thalassemia.
References
- Harteveld CL, Higgs DR. α-thalassaemia. Orphanet J Rare Dis. 2010; 5: 13. doi: 10.1186/1750-1172-5-13
- Higgs DR. The Molecular Basis of α-Thalassemia. Cold Spring Harb Perspect Med. 2013; 3(1): a011718. doi: 10.1101/cshperspect.a011718
- Weatherall DJ. Phenotype-genotype relationships in monogenic disease: lessons from the thalassaemias. Nat Rev Genet. 2001; 2(4): 245–255. doi: 10.1038/35066048
- Velasco-Rodríguez D, Blas C, Alonso-Domínguez JM, et al. Cut-Off Values of Hematologic Parameters to Predict the Number of Alpha Genes Deleted in Subjects with Deletional Alpha Thalassemia. Int J Mol Sci. 2017; 18(12): 2707. doi: 10.3390/ijms18122707
- Vayá A, Suescun M, Hernández JL, Pérez ML, Palanca S, Laiz B. Rheological red blood cell behaviour in minor α-thalassaemia carriers. Clin Hemorheol Microcirc. 2011; 48(4): 241–246. doi: 10.3233/CH-2011-1416
- Chong SS, Boehm CD, Higgs DR, Cutting GR. Single-tube multiplexPCR screen for common deletional determinants of alphathalassemia. Blood. 2000; 95(1): 360–362.
- Tan AS, Quah TC, Low PS, Chong SS. A rapid and reliable 7-deletion multiplex polymerase chain reaction assay for α-thalassemia. Blood. 2001; 98(1): 250–251. doi: 10.1182/blood.v98.1.250
- Stephens AD, Colah R, Fucharoen S, et al. ICSH recommendations for assessing automated high-performance liquid chromatography and capillary electrophoresis equipment for the quantitation of HbA2. Int J Lab Hematol. 2015; 37(5): 577–582. doi: 10.1111/ijlh.12413
- Higgs DR, Vickers MA, Wilkie AO, Pretorius IM, Jarman AP, Weatherall DJ. A review of the molecular genetics of the human α-globin gene cluster. Blood. 1989; 73: 1081–1104.
- Higgs DR, Weatherall DJ. The alpha thalassemias. Cell Mol Life Sci. 2009; 66: 1154–1162. doi: 10.1007/s00018-008-8529-9
- Old J, Harteveld CL, Traeger-Synodinos J, Petrou M, Angastiniotis M, Galanello R. Prevention of Thalassaemias and Other Haemoglobin Disorders. Volume 2: Laboratory Protocols. 2nd edition. Nicosia (Cyprus): Thalassaemia International Federation; 2012.
- Weatherall DJ. The definition and epidemiology of non-transfusiondependent thalassemia. Blood Rev. 2012; 26: S3–S6. doi: 10.1016/S0268-960X(12)70003-6
- Kalle Kwaifa I, Lai MI, Md Noor S. Non-deletional alpha thalassaemia: a review. Orphanet J Rare Dis. 2020; 15(1): 166.
- Azma RZ, Othman A, Azman N, et al. Co-inheritance of compound heterozygous Hb Constant Spring and a single -alpha(3.7) gene deletion with heterozygous deltabetathalassaemia: a diagnostic challenge. Malays J Pathol. 2012; 34(1): 57–62.
- Turbiville D, Du X, Yo J, Jana BR, Dong J. Iron Overload in an HFE Heterozygous Carrier: A Case Report and Literature Review. Lab Med. 2019; 50(2): 212–217. doi: 10.1093/labmed/lmy065
- Harteveld CL, Kriek M, Bijlsma EK, et al. Refinement of the genetic cause of ATR-16. Hum Genet. 2007; 122(3-4): 283–292. doi: 10.1007/s00439-007-0399-y
