



Research Article
Studies on the Role of hTERT – a Telomerase Regulator of DNA Methylation in Cervical Cancer
1Department of Genetics, Vrije University of Amsterdam, De Boelelaan 1085-1083, 1081 HV Amsterdam, Netherlands
2Head of Genetics Department, Bhagwan Mahavir Medical Research Centre,10-1-1, Mahavir Marg, Masab Tank, Hyderabad, Telangana, India
3Consultant Hematologist & Medical Oncologist CEO, Clinsync Clinical Research, India
*Corresponding author: Kaiser Jamil, Head of Genetics Department, Bhagwan Mahavir Medical Research Centre, 10-1-1, Mahavir Marg, Masab Tank, Hyderabad, India, E-mail: kj.bmmrc@gmail.com
Received: January 10, 2020 Accepted:February 06, 2020 Published: February 13, 2020
Citation:Asimuddin M, Jamil K, Suresh SVS. Studies on the Role of hTERT – a Telomerase Regulator of DNA Methylation in Cervical Cancer. Madridge J Case Rep Stud. 2020; 4(1): 172-179. doi: 10.18689/mjcrs-1000146
Copyright: © 2020 The Author(s). This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Telomerase is a cellular reverse transcriptase which stabilizes telomere length by adding hexameric TTAGGG repeats onto the telomeric ends of chromosomes. Expression of telomerase occurs in germ cells and stem cells but not in normal (somatic) cells. However, in many tumors, telomerase is activated, which is one of the immortalization steps. In humans, telomerase expression is regulated by a telomerase subunit called hTERT. The promoter region of the gene that encodes hTERT is located in a CpG island and was shown to be regulated, at least in part, by DNA methylation. In this study, we have analyzed the methylation status of the hTERT gene in primary squamous cell carcinomas, SCC-1 and SCC-2, and HPV+ cervical tissue scrapes, moderate dyskaryosis and mild dyskaryosis. By using the bisulfite genomic sequencing assay, the region between +90 bp and +566 bp within the gene was analyzed. We found that the primary squamous cell carcinoma SCC-1, which consists of 70% carcinoma cells, was more methylated than SCC-2, which consisted of 40% carcinoma cells. However, the number of methylated CpGs in cervical scrapes classified as moderate dyskaryosis and mild dyskaryosis were higher than in SCC-2. In the present study, we demonstrated the importance of DNA methylation profile at hTERT promoter site as a promising biomarker for cervical cancer. Further, it also provides a possible mechanism into the epigenetic regulation of hTERT in various other cancer developments.
Keywords: Telomerase subunit (hTERT); CCCTC-binding factor (CTCF); Squamous cell carcinomas (SCC); DNA Methylation; CpG island.
Introduction
Telomerase is a cellular reverse transcriptase which stabilizes telomere length by adding hexameric TTAGGG repeats onto the telomeric ends of the chromosomes. Studies of the telomerase enzyme complex has revealed that the presence of two major subunits contribute to enzymatic activity: an RNA component (hTERC) that serves as a template for the polymerase activity of the enzyme and a catalytic subunit known as human telomerase reverse transcriptase (hTERT) [1]. Both hTERC and hTERT are necessary for reconstitution of telomerase activity [2,3]. While hTERT is widely expressed in embryonic cells and in adult male germ line cells, it is undetectable in normal somatic cells except in proliferative cells of renewal tissues, including hematopoietic stem cells, activated lymphocytes, basal cells of the epidermis, proliferative endometrium, and intestinal crypt cells [4,5].
By contrast, in most somatic cells telomerase activity is down-regulated. This continues as somatic cells divide until a critical minimum telomeric length is reached after which the cells undergo cellular senescence or mortality stage-1 (M1). These cells continue to divide only when certain cell cyclec checkpoint genes such as p16, pRb and p53 are not expressed [6]. The telomeres in these senescent cells become too short and enter ‘crisis’ or mortality stage-2 (M2). This represents the physiological result of critically short telomeres when cells are no longer able to protect the ends of the chromosomes, so that end-degradation and end-to-end fusion occurs and causes genomic instability and subsequent cell death. When cells are cultured in adequate conditions, the immortal cells get reactivated by the expression of telomerase, which allow them to maintain the telomereʼs length [7].
DNA methylation of CpG island-containing promoters of certain genes, such as cell cycle regulators and tumor suppressor genes is responsible for loss of expression of these genes in many cancers [8]. The 5ʼ region of the hTERT gene consists of a large ~4kb CpG island (-1800 to +2200, numbered relative to the ATG) and considering other cases, the regulation of hTERT expression may involve DNA methylation [9,10].
To begin to evaluate this possible regulatory mechanism, several groups have assessed the methylation status of the hTERT CpG islands in a variety of normal human cells and immortal and cancer cell lines [11]. By using bisulfite treatment assays Devereux et al. [12] examined the 72 CpG sites from -500 bp upstream of transcriptional start site extending to +100 bases of exon-1. They found that most of the normal human somatic cells that do not express the hTERT gene (e.g. NHF, a normal human fibroblasts and MRC-5, a fetal lung fibroblasts) have an unmethylated hTERT promoter. On the other hand, results from the same studies shows that in hTERT expressing cell lines such as CMV, an immortal fibroblast cell line and SiHa, a cervical cancer cell line, the promoter is completely methylated [11,13].
In addition to the importance of the region upstream of the transcription start site, Renaud et al. [14] reported that the proximal exonic region of the hTERT gene also plays a role in the regulation of hTERT transcription. Experiments done by this group, using reporter gene construct, showed that including exon-1, the first intron and part of exon-2 reduced hTERT promoter activity strongly and in all cell lines tested. “This exonic region also inhibited the transcriptional activity of the CMV and CDKN2A promoter, regardless of the cell type”. Therefore, the result from Renauld et al. [14] showed that the repressive effect of the hTERT exonic region is not cell or core promoter dependent. Further investigation from this group suggested that the repressive effect is due to binding of the CCCTC-binding factor CTCF, an 11-zinc finger DNA-binding factor. CTCF is a methylation-sensitive transcription repressor which binds only to unmethylated DNA and can inhibit the transcription of the gene [15].
In vitro, EMSA studies showed that CTCF binds to the proximal exonic region at two distinct sequences, one in exon1 at +4 to +39 and one in exon-2 at position +422 to 440 bp [14]. In vivo, chromatin immunoprecipication (CHIP) assays confirmed that CTCF binds to the proximal exonic region in telomerase-negative cells, but not in telomerase-positive cells [15]. These results may give an explanation for the effect of the 5ʼ gene region on hTERT expression. Although the CTCF effect appears to be cell type independent little is known about the methylation of the CTCF binding sequences in primary cervical squmaous cell carcinomas and normal cervical cells.
Cervical cancer is one of the most common cancers among women worldwide accounting for an estimated 500,000 new cases each year [16]. Epidemiologic and molecular studies show that infection with the human papilloma virus (HPV) is a major risk factor in developing cervical cancer. Some studies have reported that as high as 99.7% of cervical carcinoma cases are due to HPV [17-19].
More than 100 different HPV types have been characterized by nucleotide sequence analysis which can be divided into low risk and high risk HPV with regard to their oncogenic effect. Infection with high-risk HPV types plays a major role in the development of cervical cancer in vivo and can induce immortalization of primary human keratinocytes in vitro [20]. The most common high-risk strains are HPV-16 and HPV-18 [21,22]. These high risk groups HPVs produce two viral oncoproteins, E6 and E7, which alter the function of cellular regulatory proteins. The E6 protein cooperates with E6AP (E6-associated protein), an ubiquitin E3 ligase, which degrades p53 via an ubiquitin-dependent pathway [23]. The E7 protein interacts with the retinoblastoma protein pRb and the E2F transcription factor which controls the expression of genes involved in cell-cycle progression [24,25]. As a result of these and other activities of the E6/E7 proteins, keratinocytes reactivate and express the hTERT gene.
Telomerase activity has been detected in advanced stages of cervical cancer cells [11] suggesting that telomerase activation is an important step towards the development of cervical cancer. To better understand these cervical carcinogenesis characters, it is important to identify the regulation of hTERT expression. The methylation status of hTERT has been described as an important component in the regulation of hTERT expression. Using bisulfite genomic sequencing, we analyzed the methylation status of the hTERT promoter downstream of the transcription start site in primary squmaous cell carcinomas (SCC) and normal but HPV infected cervical cells.
Material and Methods
Collection of specimens
For the usage of the clinical materials for research purposes, prior patient consent and approval from the Institutional Research Ethics Committee were obtained. Vaginal swabs were collected from more than 50 women attending the Department of Obstetrics and Gynecology, Mahavir Hospital and Research centre for routine checkup and also samples were collected from free health camps of the hospital premises. We selected two types of primary cervical Squamous cell carcinomas (SCC) for our investigation. In type-I, we had about 70% SCC samples and in type-II, we had 40% SCC samples. Out of these two different squamous cell carcinomas, we have selected one from each type and named as SCC-1 (type I) and SCC-2 (type IV), consisting of approximately 70% and 40% carcinoma cells for our study, respectively [11].
DNA isolation
DNA of squamous cell carcinomas was isolated by phenol/chloroform extraction as described by [26]. DNA of cervical scrapes was isolated using the High Pure PCR Template Preparation Kit (HPPTP) (Roche Applied Sciences, Indianapolis, IN).
Bisulfite treatment of DNA
The sodium bisulfite modification was done for genomic DNA isolated from different cell lines used in the experiment was carried out and provided by Li et al. [27]. For PCR amplification of the hTERT promoter regions after bisulfite modification, primers were designed to amplify the region from +90 to +350 relative to the ATG:
(hTERT-1: 5ʼ TTTGGGGTTTTAGGGTTGG-3ʼ) and
(hTERT-2: 5ʼAACCACCAACTCCTTCAAA -3ʼ)
and the region from nucleotide position +320 to +566
(hTERT-3: 5ʼ-GTAGGTGTTTTGTTTGAAGGA- 3ʼ) and
(hTERT-4: 5ʼ-TACCAACAAATAAA CCAACAC- 3ʼ).
The regions were amplified by using the MRC- Holland Master Mix (Hudsonstraat 68, 1057SN Amsterdam, The Netherlands) in a final volume of 25 µl containing 25 ng of DNA. The PCR conditions were: 95°C for 4 min, and 40 cycles, each consisting of 95°C for 1 min, 60°C for 1 min, and 72°C for 1 min. PCR fragments were checked for the correct size and the amount on a 1% agarose gel.
Cloning of PCR fragments
Before the PCR fragments were cloned into the pGEM T-easy vector (Promega), an “A” was added to the 3ʼ ends. For that, 15 µl of the first PCR reaction sample was taken and mixed with 1 µl of 10x PCR buffer, 2.5 µl of dNTPS, 6 µl of water, and 0.5 µl of Taq polymerase (2 units/µl), incubated at 70°C for 30 minutes. The PCR fragments were subsequently ligated (Promega, Woods Hollow Rd. Madison, USA) into pGEM-T Easy plasmid in the following reaction mixture contained: 3 µl of 30 ng of the PCR fragment, 1 µl of pGEM-T Easy vector, 5 µl of 2x Rapid ligation buffer, 1 µl of T4 DNA ligase (3 Weiss units/µl), the ligation mixture was incubated at 16°C overnight. Before the transformation was done, the mixture was incubated at 65°C for 15 minutes.
Protocol for E.coli competent cell preparation
E.coli cells were made competent as follows: a single colony of E.coli strain, DH5α was taken with a toothpick and put into 5 ml of L-broth and grown overnight at 37°C with agitation at 200 rpm. 300 ml of L-broth was inoculated with the 5 ml overnight culture and incubated at 37°C with agitation at 200 rpm. Cell growth was monitored until it had reached an optical density of 600 nm (OD600), which took usually about 5 hours. The culture was cooled down on ice and divided in 50 ml Nalgene tubes. The cells were collected by centrifugation at 3500 rpm at 4°C for 15 minutes. Pellets were resuspended in 50 ml of ice cold 10% glycerol and cells spin down again for 15 minutes at 3500 rpm (+4°C). This step was repeated once more. Finally, the cells were resuspended in 3-4 ml of ice-cold 10% glycerol, and divided in 0.5 ml micro centrifuge tubes and used directly or frozen in liquid N2 and stored at -80°C until use.
Transformation by electroporation and PCR
Transformation was performed using 4 µl of the ligation together with 36 µl of water and mixed with 40 µl of electro competent E.coli DH5α. Electroporation was done with a Bio-Rad Gene pulserTm with the “Set Volts” settings to 1.5 kV, “Capacity” setting to 25 µFD and resistance setting to 200 Ω. 1 ml of SOC medium was added directly after the pulse and cells transferred to a 1.5 ml tube in which they were incubated at 37°C for 30 to 35 minutes. Finally the cells were spread onto 1.5% agar LB plates (per liter 10 grm Tryptone, 5 grm yeast extract, 1.0 grm Nacl), which contained 720 µg ampicillin per liter, 1600 µg isopropylthio-β-D-galactoside (IPTG) per liter, 800 µg 5-bromo-4-chloro-3-indolyl-β-D-galactosidase (X-gal) per liter. The transformants were allowed to grow overnight at 37°C. Cells from a single E. coli colony were picked with a sterile toothpick and mixed with 10 µl of water, PCR reaction was performed by taking 5 µl of colony mixture, 2.5 µl of 10x reaction buffer, 2.5 µl of 10x dNTPS, 1 µl (20 pmol) of M13 forward and M13 reverse primer, 1 µl of Taq polymerase (2 units/µl), and 13 µl of water. The PCR conditions were: 95°C for 3 min, and 40 cycles each consisting of 95°C for 45 seconds, 56°C for 45 seconds, and 70°C for 1 min. PCR products were examined on a 1% agarose gel.
Sequence analysis
Sequence analysis of each cell line and each amplified region of the hTERT gene, for this 20 cloned PCR fragments were sequenced by using the Big Dye Terminator Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA). For this 0.3 µl of cloned PCR product was mixed with 4.7 µl of water, 3 µl of 2.5x reaction buffer, 1 µl (5 pmol) of M13 forward or with M13 reverse primer, and 1 µl of premix. The reaction was as follows: 94°C for 3 minutes, followed by 25 cycles: each 94°C for 45 seconds, 56°C for 90 seconds, 50°C for 90 seconds. The DNA fragments were purified by adding 40 µl of water, 5 µl of 3M NaAc, 1 µl of Glycogen and 135 µl of 96% EtOH. This mixture was kept on ice for 15 minutes, spin for 25-30 minutes at 13000 rpm. The pellet was dissolved in 40 µl of water. The sequence was determined on the ABI Prism 3100 Avant Genetic Analyzer (Applied Biosystems). The nucleotide sequences were analyzed with the DNA analysis program Vector NTI Advance-10 (Invitrogen, Merelbeke, Belgium). Sequences were assembled by using Contig Express, and AlignX was used to align wild type and bisulfite converted nucleotide sequences.
Results
hTERT promoter CpGs methylation map
We examined the methylation pattern of the hTERT gene downstream of the transcription start site in primary squamous cell carcinomas (SCC) and HPV positive but normal cervical tissue. Primary squamous SCC-1 and SCC-2 tissues contained approximately 70% and 40% carcinoma cells, respectively. Cervical scrapes were classified as mild and moderate dyskaryosis, respectively. Methylated CpGs in the region between +90 to +566, relative to translation start site were mapped by bisulfite genomic sequencing. The amplification of this region was performed by using different two different primer sets: hTERT-1 and hTERT-2 for the region between +90 to +350, which contains 27 CpGs. hTERT-3 and hTERT- 4 for the region between +320 to +566 which contained 28 CpGs.
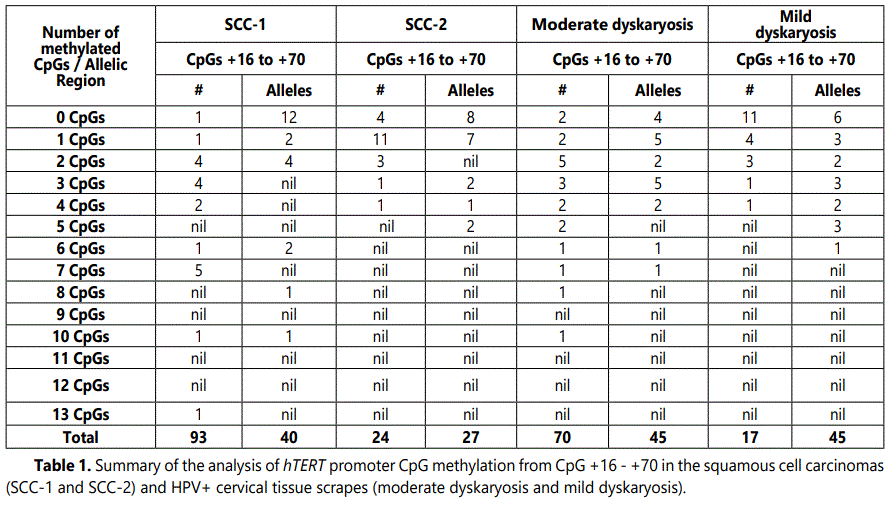
PCR fragments were analyzed on a 1% agarose gel, containing ethidium bromide (Figure 1). The lanes from 1-20 are from individual clones and (M) indicates the DNA marker. The bands at about ~500 bp contain the cloned hTERT gene fragments. The lanes showing shorter fragments represent empty PGEM-TEasy vectors. The same method was applied for the region between +350 to +566, which contains ~247 bp fragment of hTERT gene. For each region and for each cell line about 20 individual inserts were sequenced. For further investigation, we mapped the region downstream to the ATG which contains two CTCF binding sites (Figure 2a). The first CTCF-1 site is within the exon-1 between nucleotide position +4 to +39 and the second CTCF-2 site is within the exon-2 between +422 to +440 base pair relative to ATG. In this study, we analyzed the methylation status of the CpGs between nucleotide positions +90 to +566 relative to the ATG, which contains the second putative CTCF-2 binding site.
hTERT CpG methylation in primary squamous cell carcinomas (SCC)
We analyzed the hTERT 5ʼ gene region, which may contain promoter cis-acting elements, for CpG methylation in 20 cloned PCR fragments obtained after bisulfite treatment of genomic DNA. Comparison of the 20 sequences revealed that they were all different indicating that the fragments are derived from different alleles and shows the epigenetic variation of hTERT alleles. Since the non-CpG cytosines were all changed into thymines, it indicates that the bisulfite conversions were complete. The sequence differences at the CpG sites are therefore assumed to be due to differences in CpG methylation in the genomic DNA. Of the squamous cell carcinomas SCC-1 and SCC-2, SCC-1 contains slightly more methylated alleles than SCC-2 (Figure 2b and 2c) which might be related to the higher percentage of carcinoma cells in SCC-1 relative to SCC-2 (70 vs. 40%). In SCC-1, the CpGs +16 - +33 CpG are less frequently methylated than the CpGs further downstream. Although it is hard to identify a pattern, in particular the CpGs +37 - +42 were more frequently methylated (30- 50% CpGs) than the CpGs +16 - +35. Note that in none of the clones CpG +19, +26 and +36 was methylated (Figure 2b).
It was surprising to find that the higher methylation in the region containing the CpGs +37/+42 in SCC-1 did not continue in the region further downstream for the CpGs +43/+70. In all SCC-1 clones, the CpGs were completely unmethylated up to CpG +50. As the CpG +16/+42 and +43/+70 regions were amplified separately it is not possible to determine which of the methylation patterns of the first region is linked or belongs to a particular pattern of the second region (Figure 2b and 2c). However, the data suggests that the methylation after CpG +42 drops dramatically irrespective of the methylation status of the more upstream CpGs. For the region +16/+42, in total 17% of the CpGs were found methylated. Although, the number of methylated CpGs in SCC-2 was lower than in SCC-1, it was interesting to see that the methylated CpGs from +16/+41 seem to be randomly distributed (Figure 2c). Surprisingly, however, in SCC-2, the CpG +42 was frequently methylated, approximately 65% of the CpGs were methylated which was higher than in SCC-1 (45%). The CpG +43/+70 region is comparable to that of SCC-1. Eight clones were completely unmethylated and seven clones contained just one methylated CpG (Figure 2c). In SCC-2 total less than 5% of the CpGs was methylated whereas in the case of SCC-1 it was 7%. We found that the distribution of methylated CpGs in the individual clones or alleles differed substantially; this may indicate that the alleles and perhaps the cells are epigenically quite different.
hTERT CpG methylation in HPV positive cervical tissue scrapes
In moderate dyskaryosis the CpG region from +16/+42 showed more methylated CpGs than mild dyskaryosis (Figure 2d). The methylations of the +43/+70 CpGs in these two samples were similar (45 methylated CpGs each out of 560 CpGs tested) (Figure 2e). However, there are some allelic differences in the pattern. In moderate dyskaryosis, the CpGs +16/+23 CpGs were more methylated than the CpGs further downstream (CpG +24/+36). The CpGs from +37/+42 were slightly more methylated (15-20% CpGs) than the CpGs +16/+23. In the moderate dyskaryosis, the CpGs +43/+49 and +56/+59 CpGs were unmethylated in contrast to the CpG region further downstream +63/+70 (Figure 2d and 2e).
In mild dyskaryosis, the region from +16/+42 containing 27 CpGs sites was studied. For the region +16/+42, 11 alleles were completely unmethylated. The quantification of the methylated CpGs in the region +16/+42, indicated that 0-3% of the CpGs were methylated in this region. The CpGs +43/+57 CpG were less frequently methylated than the CpGs further downstream. It is difficult to identify a pattern in CpG methylation in the region +58/+70. We found that in none of the clones, CpG +43, +45, +47, +49, +54, +59, and +67 were methylated (Figure 2e).
DNA methylation patterns in cell lines
We next determined the relationship between DNA methylation patterns in each individual cell line. The primary squamous cell carcinoma SCC-1 showed the highest level of methylated CpGs from +16/+70. Approximately 133 CpGs were methylated out of 1100 CpGs tested (Table 1). In contrast to SCC-1, the HPV infected non-carcinomal cervical tissue with moderate dyskaryosis showed a similar methylation level: 115 out of 1100 CpGs tested. It was also interesting to note that mild dyskaryosis and SCC-2 had comparable overall methylation levels: 62 and 51 methylated CpGs out of 1100, respectively. The biggest differences were found in the region containing the CpGs +16/+70 of mild dyskaryosis and SCC-2.
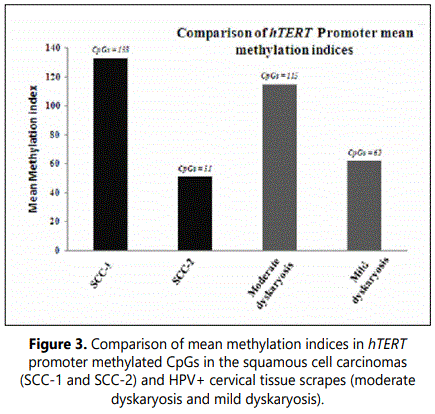
The Quantitative analysis of CpGs methylation in primary squamous cell carcinomas (SCC-1 and SCC-2) showed that SCC-1 contained slightly more methylated alleles than SCC-2 which might be related to the higher percentage of carcinoma cells in SCC-1 relative to SCC-2 (70 vs. 40%) (Figure 3). Similarly, in HPV infected tissue scrapes, moderate dyskaryosis showed more methylated alleles than in mild dyskaryosis.
Discussion
A major mechanism to regulate telomerase activity is the transcriptional control of hTERT, the catalytic subunit of the human telomerase. The regulation of the hTERT gene is complex and appears to occur through different pathways. The presence of a dense CpGs rich region in the promoter of hTERT suggested a possible role for methylation - controlled expression in normal and cancer cells [28].
In the case of cervical cancer, an aberrant methylation pattern was found during the multistage pathogenesis with an increasing trend to methylation associated with increasing pathological changes [29]. Promoter hypermethylation of various genes is a frequent epigenetic event in cervical carcinomas [30-33]. One study described the hypermethylation of the hTERT promoter region in the SiHa cervical cancer cell line [34]. The region from 500 bp upstream of transcription start site extending into exon-1 was densely methylated. Despite this methylation pattern, hTERT is expressed, indicating that methylation itself is not necessarily associated with transcriptional repression of hTERT. Given the involvement of transcriptional activators and repressors, whose binding to their cis-acting sites in the hTERT promoter may be affected by CpG methylation, it might be relevant which sequences are methylated and which are not. The information about the effects of methylation on hTERT expression in cervical cancer is in part derived from cell lines and HPV transformed primary keratinocytes [11].
The carcinomas SCC-1 and SCC-2 that were examined in this study contained approximately 70% and 40% carcinoma cells, respectively. The CpG methylation of the region downstream of the transcription start site in SCC-1 was more than two-fold higher than in SCC-2, 12% and 5% of the ~1100 CpGs tested, respectively. This difference reflects approximately the difference in the fraction of carcinoma cells in the two samples and it is therefore tempting to link the methylation to the process of tumorigenesis. However, when compared to the cervical scrapes that were analyzed, the differences were minimum. The fraction of methylated CpGs (10%) in the cervical scrape such as moderate dyskaryosis, was only slightly less than that of SCC-1 cells (12%). The cervical scrape, in the case of mild dyskaryosis was similar to that of SCC-2 (6% and 5% methylated CpGs, respectively).
The expression of hTERT in the specimens examined in the carcinomas appears to be reactivated, if methylated. It is expected that the ratio of the number of methylated to unmethylated alleles in the SCC-1 sample would be higher than in the SCC-2 sample, because SCC-1 contains almost twice as many carcinoma cells than SCC-2. In line with the heterogeneity of the sample, several clones/alleles contained very few, if any, methylated CpGs. The cellular heterogeneity of samples is only fully revealed by bisulfite sequencing and by examining methylation by MS-PCR. For each region and sample, 20 different clones/alleles were analyzed to detect patterns that can be correlated with hTERT expression and number of carcinoma cells. If hTERT is activated in the carcinoma cells it will be interesting to see to what extent it is activated in, for example, SCC-1 methylation is about 3-fold lower than in the hTERT-expressing HPV transformed keratinocytes and almost 6–fold lower than in the CaSki and SiHa cells [11].
As previously mentioned by Ramlee et al. [35], it might be that hTERT expression in normal cell is repressed by a mechanism which is no longer functional in tumors. The simplest model of transcriptional repression of hTERT in normal cells is to invoke a transcriptional repressor which is unable to work in cancer cell. According to Renaud et al. [15] the CTCF binding sites are expected to be methylated in the HPV transformed cells and the cervical cancer cell line so that there is a repression of hTERT expression. Our results show that CTCF binding region between CpG sites +54 to +56 were less methylated than the surrounding CpG sites. This was also observed in various human cancer cell lines such as Caski, SiHa and Hela cells including primary tumors and cervical carcinoma cells (Hypomethylated CpG around the transcription start site enables TERT expression and HPV16 E6 regulates TERT methylation in cervical cancer cells [11,36]. Moreover study published by Guilleret et al. [37] showed high level of methylation at both CTCF binding sites in Hela cells, which could not be confirmed by most of the other studies. This may be due to different Hela-cells subclones that were used [38,39]. However, the mechanism remains unknown about how CTCF is able to bind on hTERT and represses transcription when flanking sequences are severely methylated.
Conclusions
It is concluded from this study that the expression and CpG methylation pattern for hTERT do not constitute a contradiction to the well-established association of promoter region methylation with gene silencing as suggested by some previous reports. Our finding suggests that there may be a relation between the hTERT methylation status and pattern, or the process of tumorigenesis. The results with the primary tumors SCC-1 and SCC-2 suggest that methylation of hTERT occurs gradually and it might be a late event in tumorigenesis. The results of the HPV positive specimens suggests that this gradual increase may have already started after the infection with HPV, in line with observations with HPV transformed primary keratinocytes in which the hTERT regions examined in this study were methylated. Our study revealed that the detection of DNA methylation pattern of hTERT gene may provide an attractive new biomarker for the detection of cervical cancer.
Conflict of Interest
None
Acknowledgements
We are grateful to Mahavir Hospital and Research Centre for their encouragement and Indo American Cancer Institute for the Samples. We are also thankful to all the individuals who were part of this study group and for their consent.
References
- MacNeil DE, Bensoussan HJ, Autexier C. Telomerase Regulation from Beginning to the End. Genes (Basel). 2016; 7(9): E64. doi: 10.3390/genes7090064
- Giardini MA, Segatto M, da Silva MS, Nunes VS, Cano MI. Telomere and telomerase biology. Prog Mol Biol Transl Sci. 2014; 125: 1-40. doi: 10.1016/B978-0-12-397898-1.00001-3
- Martinez P, Blasco MA. Replicating through telomeres: a means to an end. Trends Biochem Sci. 2015; 40(9): 504-515. doi: 10.1016/j.tibs.2015.06.003
- Brey EM, Greisler HP. Telomerase expression in somatic cells. Lancet. 2005; 365(9477): 2068-2069. doi: 10.1016/S0140-6736(05)66715-3
- Brazvan B, Ebrahimi-Kalan A, Velaei K, et al. Telomerase activity and telomere on stem progeny senescence. Biomed Pharmacother. 2018; 102: 9-17. doi: 10.1016/j.biopha.2018.02.073
- Asimuddin M, Jamil K. Insight into the DNA repairs mechanism operating during cell cycle checkpoints in eukaryotic cells. Biology and Medicine. 2012; 4(4): 147-166.
- Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011; 144(5): 646-674. doi: 10.1016/j.cell.2011.02.013
- Jamil K. Cancer communications for the development of personalized medicine. J Solid Tumors. 2012; 2: 3. doi: 10.5430/jst.v2n2p1
- Zhu J, Zhao Y, Wang S. Chromatin and epigenetic regulation of the telomerase reverse transcriptase gene. Protein Cell. 2010; 1(1): 22-32. doi: 10.1007/s13238-010-0014-1
- Ramlee MK, Wang J, Toh WX, Li S. Transcription regulation of the human telomerase reverse transcriptase (hTERT) gene. Genes (Basel). 2016; 7(8): E50. doi: 10.3390/genes7080050
- de Wilde J, Kooter JM, Overmeer RM, et al. hTERT promoter activity and CpG methylation in HPV-induced carcinogenesis. BMC Cancer. 2010; 10: 271. doi: 10.1186/1471-2407-10-271
- Devereux TR, Horikawa I, Anna CH, Annab LA, Afshari CA, Barrett JC. DNA methylation analysis of the promoter region of the human telomerase reverse transcriptase (hTERT) gene. Cancer Res. 1999; 59(24): 6087-6090.
- Chung J, Khadka P, Chung IK. Nuclear import of hTERT requires a bipartite nuclear localization signal and Akt-mediated phosphorylation. J Cell Sci. 2012; 125: 2684-2697. doi: 10.1242/jcs.099267
- Renaud S, Bosman FT, Benhattar J. Implication of the exon region in the regulation of the human telomerase reverse transcriptase gene promoter. Biochem Biophys Res Commun. 2003; 300(1): 47-54. doi: 10.1016/s0006-291x(02)02775-4
- Renaud S, Loukinov D, Bosman FT, Lobanenkov V, Benhattar J . CTCF binds the proximal exonic region of hTERT and inhibits its transcription. Nucleic Acids Res. 2005; 33(21): 6850-6860. doi: 10.1093/nar/gki989
- Muñoz N, Bosch FX, de Sanjosé S, et al. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med. 2003; 348(6): 518-527. doi: 10.1056/NEJMoa021641
- Clifford GM, Smith JS, Aguado T, Franceschi S. Comparison of HPV type distribution in high-grade cervical lesions and cervical cancer: a meta-analysis. Br J Cancer. 2003; 89(1): 101-105. doi: 10.1038/sj.bjc.6601024
- Rambout L, Hopkins L, Hutton B, Fergusson D. Prophylactic vaccination against human papilloma virus infection and disease in women: a systematic review of randomized controlled trials. CMAJ. 2007; 177(5): 469-479. doi: 10.1503/cmaj.070948
- Elmi AA, Bansal D, Acharya A, et al. Human Papillomavirus (HPV) Infection: Molecular Epidemiology, Genotyping, Seroprevalence and Associated Risk Factors among Arab Women in Qatar. PLoS One. 2017; 12(1): e0169197. doi: 10.1371/journal.pone.0169197
- Liu X, Disbrow GL, Yuan H, Tomaic V, Schlegel R. Myc and human papillomavirus type 16 E7 genes cooperate to immortalize human keratinocytes. J Virol. 2007; 81(22): 12689-12695. doi: 10.1128/JVI.00669-07
- Bello BD, Spinillo A, Alberizz P, et al. Cervical infections by multiple human papillomavirus (HPV) genotypes: Prevalence and impact on the risk of precancerous epithelial lesions. J Med Virol. 2009; 81(4): 703-712. doi: 10.1002/jmv.21429
- Li N, Franceschi S, Howell-Jones R, Snijders PJ, Clifford GM. Human papillomavirus type distribution in 30,848 invasive cervical cancers worldwide: Variation by geographical region, histological type and year of publication. Int J Cancer. 2011; 128(4): 927-935. doi: 10.1002/ijc.25396
- Brimer N, Drews CM, Vande Pol SB. Association of papillomavirus E6 proteins with either MAML1 or E6AP clusters E6 proteins by structure, function, and evolutionary relatedness. PLoS Pathog. 2017; 13(12): e1006781. doi: 10.1371/journal.ppat.1006781
- Davey NE, Travé G, Gibson TJ. How viruses hijack cell regulation. Trends Biochem Sci. 2011; 36(3): 159-169. doi: 10.1016/j.tibs.2010.10.002
- Klingelhutz AJ, Roman A. Cellular transformation by human papillomaviruses: lessons learned by comparing high- and low-risk viruses. Virology. 2012; 424(2): 77-98. doi: 10.1016/j.virol.2011.12.018
- Mirmomeni MH, Sajjadi Majd S, Sisakhtnezhad S, Doranegard F. Comparison of the Three Methods for DNA Extraction from Paraffin-Embedded Tissues. Journal of Biological Sciences. 2010; 10(3): 261-266. doi: 10.3923/jbs.2010.261.266
- Li Y, Tollefsbol TO. DNA methylation detection: Bisulfite genomic sequencing analysis. Methods Mol Biol. 2011; 791: 11-21. doi: 10.1007/978-1-61779-316-5_2
- Smogorzewska A, de Lange T. Regulation of telomerase by telomeric proteins. Annu Rev Biochem. 2004; 73: 177-208. doi: 10.1146/annurev.biochem.73.071403.160049
- Laird PW. Cancer epigenetics. Hum Mol Genet. 2005; 14: R65-R76. doi: 10.1093/hmg/ddi113
- Virmani AK, Muller C, Rathi A, Zoechbauer-Mueller S, Mathis M, Gazdar AF. Aberrant methylation during cervical carcinogenesis. Clin Cancer Res. 2001; 7(3): 584-589.
- Widschwendter A, Müller HM, Fiegl H. DNA methylation in serum and tumors of cervical cancer patients. Clin Cancer Res. 2004; 10(2): 565-571. doi: 10.1158/1078-0432.ccr-0825-03
- Steenbergen RD, Kramer D, Braakhuis BJ, et al. TSLC1 gene silencing in cervical cancer cell lines and cervical neoplasia. J Natl Cancer Inst. 2004; 96(4): 305. doi: 10.1093/jnci/djh031
- Dueñas-González A, Lizano M, Candelaria M, Cetina L, Arce C, Cervera E. Epigenetics of cervical cancer: An overview and therapeutic perspectives. Mol Cancer. 2005; 4: 38. doi: 10.1186/1476-4598-4-38
- Annunziata C, Pezzuto F, Greggi S, et al. Distinct profiles of TERT promoter mutations and telomerase expression in head and neck cancer and cervical carcinoma. Int J Cancer. 2018; 143(5): 1153-1161. doi: 10.1002/ijc.31412
- Ramlee MK, Wang J, Toh WX, Li S. Transcription regulation of the human telomerase reverse transcriptase (hTERT) gene. Genes (Basel). 2016; 7(8): E50. doi: 10.3390/genes7080050
- Jiang J, Zhao LJ, Zhao C, et al. Hypomethylated CpG around the transcription start site enables TERT expression and HPV16 E6 regulates TERT methylation in cervical cancer cells. Gynecol Oncol. 2012; 124(3): 534-541. doi: 10.1016/j.ygyno.2011.11.023
- Guilleret I, Yan P, Grange F, Braunschweig R, Bosman FT, Benhattar J. Hypermethylation of the human telomerase catalytic subunit (hTERT) gene correlates with telomerase activity. Int J Cancer. 2002; 101(4): 335-341. doi: 10.1002/ijc.10593
- Renaud S, Loukinov D, Abdullaev Z, et al. Dual role of DNA methylation inside and outside of CTCF-binding regions in the transcriptional regulation of the telomerase hTERT gene. Nucleic Acids Res. 2007; 35(4): 1245-1256. doi: 10.1093/nar/gkl1125
- Lucey BP, Nelson-Rees WA, Hutchins GM. Henrietta Lacks, HeLa cells, and cell culture contamination. Arch Pathol Lab Med. 2009; 133(9): 1463-1467. doi: 10.1043/1543-2165-133.9.1463
