

Research Article
Whole transcriptome sequencing and analysis of gastrula embryos of Marsupenaeus Japonicus (Bate, 1888)
1 Ministry of Education Key Laboratory of Marine Genetics and Breeding, College of Marine Life Sciences, Ocean University of China, Qingdao 266003, China
2Qingdao No.2 Middle School of Shandong Province, Qingdao 266061, China
3Institute of Evolution and Marine Biodiversity, Ocean University of China, Qingdao 266003, China
*Corresponding author: Huarong Guo, Ocean University of China, China, Tel.: +86 532 18854261970, Email: huarongguo@ouc.edu.cn
Received: September 21, 2016 Accepted: March 30, 2017 Published: April 5, 2017
Citation: Li P, Wang Y, Wang Y, Guo H. Whole transcriptome sequencing and analysis of gastrula embryos of Marsupenaeus Japonicus (Bate, 1888). Madridge J Aquac Res Dev. 2017; 1(1): 1-7. doi: 10.18689/mjard-1000101
Copyright: © 2017 The Author(s). This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Background:
Penaeid shrimp Marsupenaeus japonicus is an economically important and widely farmed aquaculture species in China. However, information on its functional genomics or transcriptome is quite limited. Especially the whole transcriptome data from the earlystage embryos of M. japonicus remains unavailable. To discover growth-related genes involved in embryonic development, we for the first time sequenced and analyzed a whole transcriptome of gastrula embryos of M. japonicus.
Results:
In the present study, a total of 108,320,302 high-quality reads (approximately 10.84 Gb) were generated and 46,661 unigenes with an average length of 910 bp were successfully assembled and using Trinity software. After BLAST against Nr, Nt, Swissprot, KO, KOG, GO and Pfam database, among the assembled unigenes 14,399 were classified into 49 GO functional terms, 8,290 unigenes were divided into 26 KOG groups and 5, 979 unigenes were annotated into 244 KEGG pathways. On account of the GO and KEGG annotations, a total of 37 growth-related genes were identified.
Conclusion: The disclosure of gastrula embryo transcriptome dataset of this M. japonicus will provide a valuable resource for future works on the mechanism of embryonic development, identification of functional genes and genetic manipulation in penaeid shrimps.
Keywords: Shrimp; Marsupenaeus japonicus; Transcriptome; gastrula embryo; growthrelated gene
Abbreviations
Nr: Non-redundant protein;
Nt: Non-redundant nucleotide;
KEGG: Kyoto Encyclopedia of Genes and Genomes;
KO: KEGG Orthology database;
KOG: euKaryotic Ortholog Groups;
GO: Gene Ontology;
Pfam: Protein family;
BLAST: Basic Local Alignment Search Tool;
EST: Expressed Sequence Tags;
NGS: Next-Generation Sequencing;
RNA-Seq: RNA Sequencing;
WSSV: white spot syndrome virus;
TSV: Taura Syndrome Virus;
CDS: coding sequence;
SNP: Single Nucleotide Polymorphism;
RPKM: Reads Per Kilo bases per Million mapped Reads.
Introduction
Kuruma shrimp Marsupenaeus japonicus, belonging to the subphylum Crustacea of Arthropoda, is an economically important and widely farmed aquaculture species in China [1,2]. The high nutritional value, rapid growth feature and key role in evolution of M. japonicus not only made it beneficial for aquaculture industry but also get the attention of scientist to study more about its development, breeding and genetics [3,4]. Although the whole genome sequencing and assembling of penaeid shrimp are still not finished up to date due to the extremely high proportion of repeat sequences (private communication), several methods like cDNA library construction, microarray and expressed sequence tags (EST) have been used to analyze the transcriptomes of penaeid shrimps, which can shed a new light on the molecular mechanisms of many biological processes, especially the responses of shrimps to environmental stress and pathogenic infections[5-10].
During the past decade, the next-generation sequencing (NGS) techniques of Solexa (Illumina), 454 (Roche) and SOLiD (ABI) were rapidly developed one after another and widely applied in the genome and transcriptome analysis in a highthroughput, massively parallel and low cost manner[11,12]. RNA Sequencing (RNA-Seq) is a NGS-based method that allows the entire transcripts to be surveyed in a very highthroughput and quantitative manner[13,14]. Recently developed Trinity software makes it possible to efficiently assemble reads without a reference genome[15]. Both RNASeq and Trinity have greatly facilitated the transcriptome analysis in penaeid shrimps[16-28]. Most of these works focused on pacific white shrimp Litopenaeus vannamei[16-27]. For example, the transcriptomes of early embryo (mixtures of zygote, blastula, gastrula, limb bud embryo and larva in membrane)[16], nauplius[16], zoea[16], mysis[16], postlarvae[16] and 20-d larvae[17] of L. vannamei have been examined, and improved the current understanding of the molecular mechanisms of the physiological changes during shrimp metamorphosis, especially histogenesis, diet transition[18], muscle development and exoskeleton reconstruction. Gao et al. further analyzed the transcriptomic changes throughout the molting cycle of L. vannamei and got new insights into the molecular mechanisms of molting[19]. The transcriptomic changes of gills, hepatopancreas or hemocytes of L. vannamei in response to the challenge of white spot syndrome virus (WSSV) or Taura Syndrome Virus (TSV) had also been widely investigated and provided valuable information on the host-virus interaction, and a lot of immune-related genes responding to virus infection were discovered, too[20-24]. In addition, effects of nitrite exposure on the transcriptome of L. vannamei [25], and discovery of horizontally transferred genes [26] and single nucleotide polymorphism(SNP) [27] from the genome of L. vannamei were also carried out by transcriptome analysis. In contrast, Fenneropenaeus chinensis was another one species in which whole transcriptome analysis has been performed except for L. vannamei[28]. However, as far as we know, no transcriptome analysis has been reported in M. japonicus.
Whole transcriptome analysis of early stage embryos of M. japonicus is of great importance for the discovery of growth-related genes and molecular mechanism of embryonic development. In the present study, we sequenced and analyzed the whole transcriptome of gastrula embryos from M. japonicus for the first time using Illumina Hiseq 2000 platform and identified many growth-related genes.
Materials and methods
Shrimp embryos and sampling
After spawning, fertilized eggs of M. japonicus were collected from a local shrimp breeding farm (Wang-gezhuang, Qingdao, China), transferred to laboratory immediately and maintained in aerated seawater (26-28°C). Then the embryos at gastrula stage were sampled at 6-7 h post spawning and washed adequately first by 0.22 mm filtersterilized sea water and then by phosphate buffered solution (PBS)[29]. After suspended in TransZolTM reagent (TransGen Biotech, China), the samples were rapidly frozen and stored at -80°C for RNA isolation.
RNA extraction and Illumina sequencing
Total RNAs were isolated from the gastrula embryos of M. japonicus using TransZolTM according to the manufacturer's instructions. The degradation of RNA and contamination of DNA and protein were checked on 1% agarose gel electrophoresis. The purity, concentration and integrity of the isolated RNAs were monitored with the NanoPhotometer® spectrophotometer (IMPLEN, CA, USA), Qubit® 2.0 Flurometer (Life Technologies, CA, USA) and Bioanalyzer 2100 (Agilent Technologies, CA, USA), respectively. Then the total RNAs were delivered to Novogene Company (China) for the construction of cDNA library and Illumina sequencing.
A total of 3 µg total RNAs was added to generate sequencing libraries using Illumina TruSeqTM RNA Sample Preparation Kit (Illumian, San Diego, USA) following manufacturer's instructions. Briefly, the total mRNAs were purified from the total RNAs through poly-T oligo-attached magnetic beads and then Illumina-proprietary fragmentation buffer was added to carry out the fragmentation of mRNAs under elevated temperature. First strand cDNA synthesis was performed using random oligonucleotides and SuperScript II, and second strand cDNA was subsequently synthesized using DNA Polymerase and RNase H. After being purified with AMPure XP system (Beckman Coulter, Beverly, USA), the double-strand cDNAs were subjected to end repair and addition of A and adapter. The cDNA fragments around 200 bp in length were first pooled using AMPure XP beads and then the DNA fragments with adaptors on both ends were further selectively enriched using Illumina PCR Primer Cocktail in a 10 cycle PCR reaction. In the end, based on Illumina HiSeq 2000 platform, the cDNA library of the gastrula embryo transcriptome were sequenced by Paired-End sequencing method.
Trinity assembly and gene functional annotation
All the raw reads generated from the Illumina HiSeq 2000 sequencer were further filtered to obtain clean reads by removing adapters and low quality reads containing polyN(>10%). After that, transcripts were obtained by assembling the clean reads using Trinity software[15] with min_kmer_cov set to 2 and all other parameters set default. All the unigenes (i.e. the longest transcript for each gene) were then blasted against databases of NCBI non-redundant protein sequences (Nr), NCBI non-redundant nucleotide sequences (Nt), Swiss-Prot (a manually annotated and reviewed protein sequence database), euKaryotic Ortholog Groups (KOG), Protein family (Pfam), Gene Ontology database (GO) and KEGG Orthology database (KO) separately for similarity search and gene functional annotation. Table 1 summarized the used software, version and parameters for the gene functional annotation in 7 databases. Based on the annotation results of Nr database and Pfam database, GO classification of unigenes was obtained using BLAST2GO program with default parameters [30]. KOG annotation of unigenes was performed using NCBI Blast version 2.2.27+ with an E-value threshold of 10-3 against KOG database [31]. KEGG metabolic pathway classification for all the assembled unigenes was determined by KEGG Automatic Annotation Server (KASS) against KO database [32].
The assembled transcriptome was further used as a reference sequence due to the absence of genomic information of penaeid shrimps. Thus all the clean reads in the gastrula transcriptome of M. japonicus were mapped to its own transcriptome dataset using RSEM software with the bowtie parameters of mismatch 2, for the read number counting for each gene [33]. RPKM (Reads Per Kilo bases per Million mapped Reads), the most common method to estimate the gene expression level, was used to balance the influence of both sequencing depth and gene length on the read number counting [34].
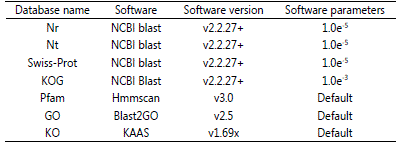
Table 1: Summary of software, version and parameters used in 7 databases
Identification of growth-related genes
In order to further analyze the mechanism of embryo development of M. japonicus, growth-related genes in the gastrula embryo transcriptome were screened out from Growth term of GO classification and Cell growth and death pathway of KEGG classification. The transcript IDs of growthrelated genes were then selected and located to the Nr, Nt, Swiss-Prot, and Pfam databases. Finally the names, the lengths, E-value and RPKM of the growth-related genes selected were summarized in a table.
Data Availability
The sequence data in this study have been deposited into the NCBI Sequence Read Archive database and the accession number is SRS1609831.
Results
Illumina sequencing and assembly of gastrula transcriptome of M. japonicus
To globally understand the expression profile of genes involved in the early embryonic development of M. japonicus, a cDNA library was constructed from the total mRNAs of the gastrula embryos and the RNA-Seq was performed using the Illumina HiSeq 2000 sequencing platform. It was found that, a total of 112,125,092 raw reads with an error percentage of 0.03% were obtained. After removal of the adapters and lowquality reads containing N (>10%), a total of 108,320,302 clean reads, with accumulated data volume of 10.84 Gb and GC percentage of 46.69%, were generated and assembled into 67,183 transcripts, ranging from 201 bp to 14,938 bp in length, with mean length of 1,216 bp and N50 length of 2,405 bp. Among the 67,183 transcripts, 45.01% (30,241) were in a length of 200-500 bp, 19.61% (13,174) were 500-1 kb, 16.73% (11,240) were 1-2 kb, 18.65% (12,528) were >2 kb. A total of 46,661 unigenes with average length of 910 bp and N50 length of 1,735 bp were obtained, of which 2,480 (52.46%) with 200-500 bp, 9,047 (19.39%) with 500-1 kb, 6,579 (15.0%) with 1-2 kb and 5,555 (11.91%) with >2 kb (Table 2 and 3).

Table 2: The size distribution of transcripts and unigenes.
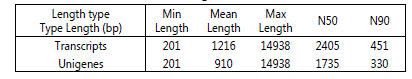
Table 3: Summary of the assembly length of transcripts and unigenes.
Notes: Min length, minimum length. Max length, maximum length. N50 and N90 define the assembly quality. N50 length is the shortest sequence length for which the collection of all contigs of that length or longer contains 50% of the given set of contigs [33]. N90 length is the length for which the collection of all contigs of that length or longer contains at least 90% of the given set of contigs.
Gene functional annotation of gastrula transcriptome of M. japonicus
All the assembled unigenes were submitted to corresponding sequence alignments for gene functional annotation with Nr, Nt, KO, KOG, GO, Swiss-Prot and Pfam databases. As shown in Table 4, among the 46,661 unigenes, 13,265 unigenes (28.42% of the total) were successfully matched in Nr database, 2,590 unigenes (5.37%) matched in Nt database, 5,979 unigenes (12.81%) matched in KO database, 11,321 unigenes (24.26%) matched in Swiss-Prot database, 13,980 unigenes (29.96%) matched in Pfam database, 14,399 unigenes (30.85%) matched in GO database and 8,290 unigenes (17.76%) matched in KOG database. A total of 1,476 unigenes (3.16%) were annotated in all databases and 17,439 unigenes (37.37%) showed matches in at least one databases.
All the successfully annotated unigenes were further applied in protein similarity search against Nr, Swiss-Prot and KO databases in priority order using BlastX software and the 0pen reading fragments (ORF) of the unigenes were determined by the optimum match results. Subsequently, the coding sequences (CDS) and amino acid sequences were confirmed based on Standard code table, resulting in 13,218 CDSs. The remaining unmatched unigenes with above-mentioned 3 protein databases were further taken into the prediction of CDS using ESTscan (v3.0.3) and translated into peptide sequences. The CDSs of 15,333 unigenes were successfully predicted covering 32.86% of the total unigenes.
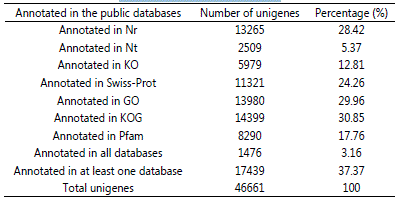
Table 4: Summary of the number and percentage of annotated genes in different databases
GO, KOG and KEGG classification
Based on the annotated protein results from Nr and Pfam databases, the software BLAST2GO was used for Gene Ontology (GO) classification and 87,158 GO term annotations corresponding to 14,399 unigenes were successfully matched and divided into 3 categories and 49 functional subgroups. Among 87,158 GO terms 40,360 (46.31%), 17,789 (20.41%) and 29,009 (32.28%) terms were mapped to the three categories of biological process, molecular function and cellular component, respectively (Fig.1). The biological process was classified into 21 groups, the top three groups were cellular process (20.94%), metabolic process (17.93%) and single-organism process (12.17%), followed by biological regulation (8.41%), regulation of biological process (8.05%), localization (6.25%), establishment of localization (6.15%) and response to stimulus (5.75%). Among the 17 GO functional groups of cellular component, cell (19.60%) and cell part (19.58%) were the top two groups, followed by organelle (11.63%), membrane (10.47%), macromolecular complex (10.0%), membrane part (9.80%) and organelle part (7.7). Among the 11 GO functional groups of molecular function, binding (45.48%), catalytic activity (32.89%) and transporter activity (7.39%) were assigned into the top three groups.
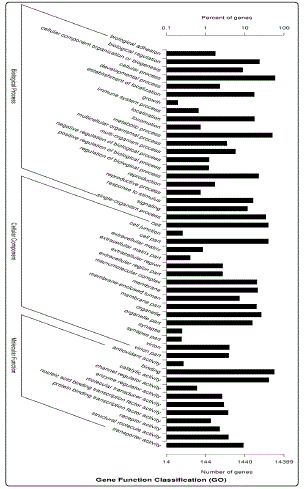
Figure 1: GO analysis of the unigenes.
As shown in Fig. 2, KOG classification was performed by NCBI Blast (v2.2.27+) with a cutoff value of E-value ≤1.0E-3 and 8,290 unigenes were searched against KOG database and classified into 26 KOG categories, of which the cluster for general functional prediction (1,802, 21.74%) was the largest group, followed by signal transduction (1,391, 16.78%), posttranslational modification, protein turnover and chaperon (834, 10.06%) and coenzyme metabolism (731, 8.82%). Taken together, a total of 1,953 (23.56%) unigenes were involved in the process of replication, transcription and translation, indicating that the transmission and expression of genetic information was a surprisingly complex event requiring massive regulatory factors. This is also in agreement with the fact of active mitosis and growth occurred in the gastrula embryos of M. japonicus.
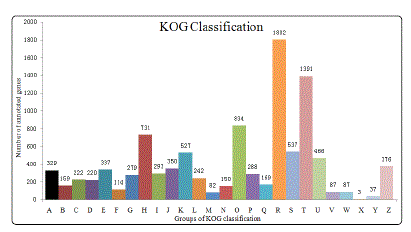
Figure 2: KOG analysis of the unigenes. A:RNA processing and modification; B: chromatin structure and dynamics;C:energy production and conversion; D: cell cycle control and mitosis; E: amino acid metabolis and transport; F: nucleotide metabolism and transport; G: carbohydrate metabolism and transporn; H:coenzyme metabolism; I: lipid metabolism; J: tranlsation; K: transcription; L: repliaction and repair; M: cell wall/membrane/envelop biogenesis; N:cell motility; O: post-translational modification, protein turnover and chaperon; P: inorganic ion transport and metabolism; Q:secondary Structure; R: general functional prediction; S: function unknown; T: signaltransduction; U:intracellular trafficing and secretion; V:defense mechanisms; W: extracellular structures; X: unamed protein;Y: nuclear structure; Z: cytoskeleton.
As shown in Fig.3, the assembled unigenes were annotated into KEGG classififcation using KASS program corresponded to KO database and a total of 5,979 unigenes were assigned to 244 metabolic pathways, which were categorized into five classess of cellular processes, environmental information processes, genetic information processes, metabolism and organismal systems. Among the KEGG annotations, more unigenes were involved in the pathway of signal transdution (614, 10.27%), followed by transport and catabolism (416, 6.96%), foling, sorting and degradation (380, 6.36%) and translation (336,5.62%). A lot of embryonic developmentrelated pathways inclding signaling pathways of Wnt, mitogen-activated protein kinase (MAPK), P13K-Akt, TGF-β etc. were successfully annotated in the KEGG database .
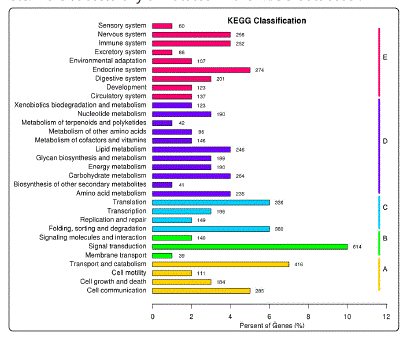
Figure 3: KEGG analysis of the unigenes. A:cellular processes; B:environmental information processes; C: genetic information processes; D:metabolism; E:organismal systems.
Identification of growth-related genes from gastrula transcriptome of Marsupenaeus Japonicus
In order to identify functional genes specifically expressed in the embryonic development, we constructed a cDNA library from gastrula embryos of M. japonicus and carried out Illumina sequencing. According to the growth term of GO annotation and cell growth and death pathway of KEGG classification, a total of 37 growth-related genes were identified and summarized in Table 5. We found that the expression abundances of these growth-related genes were quite different among them in a view of RPKM. Of them, the ring box protein and G2/mitotic-specific cyclin-B3 isoform 1 showed the highest expression level with a RPKM value of over 100, inferring their important role in the cellular proliferation and growth during the embryonic development of M. japonicus.

Table 5: Summary of the growth-related genes in the gastrula transcriptome of M. japonicus
Discussion
Penaeid shrimps are economically important and widely farmed aquaculture species in the world. However, the absence of a fully assembled reference genome has heavily hindered the works on gene identification, functional genomics and genetic manipulation in shrimps. With the emergence of the new generation of high-throughput sequencing technology, methods for transcriptome analysis have been continuously improved, especially the application of Trinity software has made it possible to efficiently assemble reads without a reference genome, thus greatly facilitated the transcriptome analysis in penaeid shrimps[15,16]. Today, many transcriptome sequencing data have been reported in F. chinensis and Macrobrachium rosenbergii [16-28]. However, no transcriptome sequencing data for the early-stage embryos of M. japonicus are available.
In the present study, we analyzed the whole transcriptome of gastrula embryos of M. japonicus using Illumina sequencing technology. A total of 108,320,302 clean reads (nearly 10.84 Gb) were generated and 67,183 transcripts and 46,661 unigenes were successfully assembled by Trinity software. The mean length of these transcripts was about 1,216 bp and 910 bp for the uingens, quite better in assembly quality than other transcriptome data reported[15-28]. The assembled unigenes were subjected to similarity search against Nr, Nt, Swiss-prot, KO, KOG, GO and Pfam databases and approximately 37.37% of the total unigenes (17,439 unigenes) were annotated in at least one database mentioned above. In contrast, 19% annotation percentage was reported in M. rosenbergii transcriptome[10], and 46.28% annotation percentage in F. chinensis transcriptome[28]. Limited information on the genome and transcriptome of crustaceans and related species, the existence of non-coding RNA in the assembled unigenes, such as long non-coding RNA with polyA structure which cannot be annotated in the public database, etc. might account for the relatively low annotation rate in penaeid shrimp transcriptomes. For the unannotated unigenes, we used Estscan (3.0.3) software to predict their CDSs and translate them into peptide sequences. Through this manual method, a total of 15,333 unigenes (32.86% of the total unigenes) were successfully predicted, which offered a rich resource for identifying novel genes. It can be expected that with more transcriptomes and genome sequencing data of shrimps disclosed, the annotation rate and identification of novel genes will be significantly increased.
To further analyze the functional annotation data, we assigned the assembled unigenes into GO, KOG and KEGG annotations, respectively. Based on the obtained GO and KEGG classifications, we identified 37 growth-related genes, which were essential to better understand the mechanism of embryonic development of penaeid shrimps and will facilitate the design of strategies to improve the breeding and aquaculture of penaeid shrimps. In a view of the RPKM value of these growth-related genes, Ring box protein and G2/mitotic-specific cyclin-B3 isoform1 were the most abundant two genes. Ring box protein has been reported to be involved in the formation of SCF (Skp-Cul1-F-box) complex, which is a kind of E3 ligase enzymes catalyzing the ubiquitination process of proteins[35]. The ubiquitination process is closedly associated with the embryonic development[36,37], suggesting a possible reason for the high expression level of Ring box protein. G2/mitotic-specific cyclin-B3 is a kind of cyclin-dependent protein kinase (cdks) which is dispensable for mitosis and thus highly needed in the embryonic development of M. japonicas [38,39].
In conclusion, a whole transcriptome of gastrula embryos of M. japonicus was successfully sequenced, de novo assembled and annotated in this study. The disclosure of this gastrula embryo transcriptome dataset and identification of 37 growthrelated genes of M. japonicus will provide us a valuable resource to facilitate our future works on the genetic manipulation and mechanisms of target biological processes interested.
ACKNOWLEDGEMENTS
This work is supported by National Natural Science Foundation of China (Grant No. 31472274 and 31172391), National High-tech R&D Program of China (863 Program; Grant No. 2012AA10A402), Fundamental Research Funds for the Central Universities of China (Grant No. 201122005), and Regional Demonstration of Marine Economy Innovative Development Project (Grant No. 12PYY001SF08).
Conflict of Interest
The authors confirm that there is no conflicts of interest regarding this manuscript.
References
- You X, Su Y, Mao Y, et al. Effect of high water temperature on mortality, immune response and viral replication of WSSV-infected Marsupenaeus japonicus juveniles and adults. Aquaculture. 2010; 305(1–4): 133–137. doi: 10.1016/j.aquaculture.2010.04.024
- Takashi K, Shuichi A, Takayuki K, et al. Hyper-expansion of large DNA segments in the genome of kuruma shrimp, Marsupenaeus japonicus. BMC Genomics. 2010; 11: 141-143. doi: 10.1186/1471-2164-11-141.
- Lyons RE, Dierens LM, Tan SH, Preston NP, Li Y. Characterization of AFLP markers associated with growthin the Kuruma prawn, Marsupenaeus japonicus, and identification of a candidate gene. Mar. Biotechnol. 2007; 9 (6): 712-721. doi: 10.1007/s10126-009-9022-4.
- Callaghan TR, Degnan BM, Sellars MJ. Expression of sex and reproductionrelated genes in Marsupenaeus japonicus. Mar. Biotechnol. 2010; 12 (6): 644-677. doi: 10.1007/s10126-009-9254-6.
- Pongsomboon S, Tang S, Boonda S, Aoki T, et al. A cDNA microarray approach for analyzing transcriptional changes in Penaeus monodon after infection by pathogens. Fish Shellfish Immun. 2011; 30(1): 439-446. doi: 10.1016/j.fsi.2010.10.015.
- Leu JH, Chen SH, Wang YB, et al. A review of the major penaeid shrimp EST studies and the construction of a shrimp transcriptome database based on the ESTs from four penaeid shrimp. Mar. Biotechnol. 2011; 13(4): 608–621. doi: 10.1007/s10126-010-9286-y.
- Pongsomboon S, Tang, Boonda S, et al. Differentially expressed genes in Penaeus monodon hemocytes following infection with yellow head virus. BMP Reports. 2008; 41(9): 670-677.
- O' Leary NA, Trent HF, Robalino J, Peck MET, McKillen DJ, Gross1 PS. Analysis of multiple tissue-specific cDNA libraries from the Pacific white leg shrimp, Litopenaeus vannamei. Integr. Comp. Biol. 2006; 46 (6): 931– 939. doi: 10.1093/icb/icl006.
- Wei J, Liu C, Zhang X, et al. Differential gene expression analysis based on expressed sequence tags( EST) from different tissues of Fenneropenaeus chinensis. J. Fish. China. 2013; 37(5): 661-671. doi: 10.3724 /SP.J.1231.2013.38371.
- Jung H, Lyons RE, Dinh H, Hurwood DA, McWilliam S, Mather PB. Transcriptomics of a Giant Freshwater Prawn (Macrobrachium rosenbergii): De Novo Assembly, Annotation and Marker Discovery. PLoS ONE.2011; 6(12): e27938. doi: 10.1371/journal.pone.0027938.
- Liu L, Li Y, Li S, et al. Comparison of next-generation sequencing system. J. Biomed Rech intentional. 2012; 2012: 251364. doi: 10.1155/2012/251364.
- Metzker ML. Sequencing technologies: the next generation. Nat. Rev. Genet. 2010; 11(1): 31-46. doi: 10.1038/nrg2626.
- Wall PK, Leebens-Mack J, Chanderbali AS, et al. Comparison of next generation sequencing technologies for transcriptome characterization. BMC Genomics. 2009; 10: 347. doi: 10.1186/1471-2164-10-347.
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009; 10: 57–63. doi: 10.1038/nrg2484.
- Grabherr MG, Haas BJ, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011; 29: 644-652. doi: 10.1038/nbt.1883.
- Wei J, Zhang X, Yu Y, Huang H, Li F, Xiang J. Comparative transcriptomic characterization of the early development in pacific white shrimp Litopenaeus vannamei. PLoS ONE. 2014; 9(9): e106201. doi: 10.1371/ journal.pone.0106201.
- Li CZ, Weng SP, Chen YG, et al. Analysis of Litopenaeus vannamei transcriptome using the next-generation DNA sequencing technique. PLoS ONE. 2012; 7(10): e47442. doi: 10.1371/journal.pone.0047442.
- Wei J, Zhang X, Yu Y, Li F, Xiang J. RNA-Seq reveals the dynamic and diverse features of digestive enzymes during early development of Pacific white shrimp Litopenaeus vannamei. Comp Biochem Phys Part D. 2014; 11: 37–44. doi: 10.1016/j.cbd.2014.07.001.
- Gao Y, Zhang X, Wei J, et al. Whole transcriptome analysis provides insights into molecular mechanisms for molting in Litopenaeus vannamei. PLoS ONE. 2015; 10(12): e0144350. doi: 10.1371/journal. pone.0144350.
- Clavero-Salas A, Sotelo-Mundo RR., Gollas-Galván T, et al. Transcriptome analysis of gills from the white shrimp Litopenaeus vannamei infected with White Spot Syndrome Virus. Fish Shellfish Immun. 2007; 23(2): 459- 472. doi: 10.1016/j.fsi.2007.01.010.
- Chen HX, Zeng DG, Chen XL, et al. Transcriptome analysis of Litopenaeus vannamei in response to white spot syndrome virus infection. PLoS ONE. 2013; 26:8(8): e73218. doi: 10.1371/journal.pone.0073218.
- Xue SX, Liu YC, Zhang YC, Sun Y, Geng XY, Sun JS. Sequencing and De Novo analysis of the hemocytes transcriptome in Litopenaeus vannamei response to White Spot Syndrome Virus infection. PLoS ONE. 2013; 8(10): e76718. doi: 10.1371/journal.pone.0076718.
- Zeng DG, Chen XL, Xie DX, et al. Transcriptome analysis of Pacific white shrimp (Litopenaeus vannamei) hepatopancreas in response to Taura Syndrome Virus (TSV) experimental infection. PLoS ONE. 2013; 8(2): e57515. doi: 10.1371/journal.pone.0057515.
- Sookruksawong S, Sun F, Liu Z, Tassanakajon A. RNA-Seq analysis reveals genes associated with resistance to Taura syndrome virus (TSV) in the Pacific white shrimp Litopenaeus vannamei. Dev. Comp. Immu. 41(4) (2013) 523–533. doi: 10.1016/j.dci.2013.07.020.
- Guo H, Ye CX, Wang AL, Xian JA, Liao SA, Miao YT. Transcriptome analysis of the pacific white shrimp Litopenaeus vannamei exposed to nitrite by RNA-seq. Fish Shellfish Immunol. 2013; 35(6): 2008-2016. doi: 10.1016/j.fsi.2013.09.019.
- Yuan J-B, Zhang X-J, Liu C-Z, Wei J-K, Li F-H, Xiang J-H. Horizontally transferred genes in the genome of Pacific white shrimp, Litopenaeus vannamei. BMC Evol. Biol. 2013, 13:165. doi: 10.1186/1471-2148-13- 165
- Yu Y, Wei J, Zhang X, et al. SNP Discovery in the transcriptome of white Pacific Shrimp Litopenaeus vannamei by next generation sequencing. PLoS ONE. 2014; 9(1):e87218. doi: 10.1371/journal.pone.0087218.
- Li SH, Zhang XJ, Sun Z, Li FH, Xiang JH. Transcriptome analysis on Chinese shrimp Fenneropenaeus chinensis during WSSV acute infection. PLoS ONE. 2013; 8(3): e58627. doi: 10.1371/journal.pone.0058627.
- Han Q, Li P, Lu X, Guo Z, Guo H. Improved primary cell culture and subculture of lymphoid organs of the greasyback shrimp Metapenaeus ensis. Aquaculture. 2013; 410-411: 101-113. doi: 10.1016/j. aquaculture.2013.06.024.
- Götz S, García-Gómez JM, Terol J, et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008; 36(10): 3420-3435. doi: 10.1093/nar/gkn176.
- Mount DW. Using the Basic Local Alignment Search Tool (BLSAT). Cold Spring Harb. Protoc. 2007. doi: 10.1101/pdb.top17.
- Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000; 28(1): 27-30. doi: 10.1093/nar/28.1.27.
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome.BMC Bioinformatics. 2011; 12:323. doi: 10.1186/1471-2105-12-323.
- Miller JR, Koren S, Sutton G. Assembly algorithms for next-generation sequencing data. Genomics. 2010; 95 (6): 315-327. doi: 10.1016/j. ygeno.2010.03.001.
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods.2008; 5(7): 621-628. doi: 10.1038/nmeth.
- Santos DN, Aguiar PH, Lobo FP, et al. Schistosoma mansoni: Heterologous complementation of a yeast null mutant by SmRbx, a protein similar to a RING box protein involved in ubiquitination. Exp Parasitol. 2007; 116(4): 440-449. doi: 10.1016/j.exppara.2007.02.012
- Jasinskiene N, Jasinskas A, Langmore JP. Embryonic regulation of histone ubiquitination in the sea urchin. Dev. Genet. 1995; 16(3): 278-290.doi: 10.1002/dvg.1020160308
- Oshima S, Turer EE, Callahan JA, et al. (2009). ABIN-1 is a ubiquitin sensor that restricts cell death and sustains embryonic development. Nature 2009; 457(7231): 906-919. doi: 10.1038/nature07575.
- Gallant P, Nigg EA. Identification of a novel vertebrate cyclin: cyclin B3 shares properties with both A- and B-type cyclins. EMBO J. 1994; 13(3): 595-605.
- Jacoba HW, Knoblich JA, Lehner CF. Drosophila Cyclin B3 is required for female fertility and is dispensable for mitosis like Cyclin B. Genes Dev. 1998; 12(23): 3741-3751.
