

Research Article
Determination of Fluoroquinolones in Milk by Ionic Liquid-Mediated two phase extraction followed by Capillary Electrophoresis Analysis
Key Laboratory of Theoretical and Computational Photochemistry, Ministry of Education, College of Chemistry, Beijing Normal University, Beijing 100875, China
*Corresponding author: Weidong Qin, Key Laboratory of Theoretical and Computational Photochemistry, Ministry of Education, College of Chemistry, Beijing Normal University, Beijing 100875, China, Fax: + 86-10-58802531, Email: qinwd@bnu.edu.cn
Received: May 2, 2018 Accepted: May 16, 2018 Published: May 23, 2018
Citation: Tian Z, Gao JJ, Qin W. Determination of Fluoroquinolones in Milk by Ionic Liquid-Mediated two phase extraction followed by Capillary Electrophoresis Analysis. Madridge J Anal Sci Instrum. 2018; 3(1): 62-67. doi: 10.18689/mjai-1000112
Copyright: © 2018 The Author(s). This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
We synthesized a surfactant ionic liquid (IL) 1-dodecyl-3-methylimidazolium bromide ([C12MIM]Br), and employed it as an additive to the dichloromethane-based liquid-liquid extraction (LLE) of five fluoroquinolones (FQs), namely, gatifloxacin, lomefloxacin, enoxacin, ofloxacin and pazufloxacin, which were subsequently analyzed with capillary electrophoresis- ultraviolet detection (CE-UV). The parameters affecting the IL-mediated LLE efficiency were investigated. It was found that addition of 2.0 g/L [C12MIM]Br to the donor phase, which was adjusted to pH 8.2 with 40 mM phosphatetetraborate, could improve the recoveries by 30%-100% for the five model FQs. Incorporated with CE-UV, the method could determine FQs in milk samples with detection limits ranging from 0.003 to 0.024 μg/mL. Our results suggest that the ILmediated LLE method is efficient, fast and easy to operate, and the combinational strategy of IL-mediated LLE-CE can be applied to highly efficient, sensitive, selective detection of trace FQ residues in complicated matrices.
Keywords: Imidazolium ionic liquid; Fluoroquinolones; Liquid-liquid extraction; Capillary electrophoresis.
Abbreviations: Fluoroquinolones, FQs; Liquid-liquid extraction, LLE; Gatifloxacin, GAT; Lomefloxacin, LOM; Enoxacin, EN; Ofloxacin, OF; Pazufloxacin, PZ; 1-dodecyl-3-methylimidazolium bromide, [C12MIM]Br; Recovery enhancement factor, REF
Introduction
Fluoroquinolones (FQs) are antibiotics for both pathogenic Gram-negative and Gram-positive bacteria and can inhibit bacterial DNA synthesis [1]. Since the 1980s, they have been widely used for the prevention and treatment of various infectious diseases in animals and human beings. However, FQs have drawn ongoing concerns owing to the possible toxic effects, allergic reactions, and development of resistant strains of bacteria. Therefore, the maximum residue limits (MRLs) for FQ residues in animalderived foods have been set by the European Union and other countries and regions [2-3] and methods for quick and accurate determination of FQs in animal-derived foods are highly needed [4].
The analysis of FQs residues in biological samples is predominately performed by high performance liquid chromatography (HPLC) [5-7]. In recent years, capillary electrophoresis (CE) has been increasingly applied to FQ determination due to its high separation efficiency and ease for automation [8-9]. However, despite the improvements in the instrumental analysis techniques, determination of FQ residues in the complex matrices of real samples is still challenging because the residual concentration of FQs in biological samples is normally very low while there are many interfering substances in the real samples. As a result, a pretreatment step is necessary before measurement to enrich the FQs and eliminate the interfering materials. Among the numerous sample preparation methods, solid-phase extraction [10-11], micro-solid phase extraction [12] and liquid-phase microextraction [13] are efficient protocols for sample clean-up and analyte enrichment. Nonetheless, those techniques are either relatively costly or sophisticated instrumentdemanding, or both. Liquid-liquid extraction (LLE) is a commonly used sample pretreatment technique for FQs, but single LLE efficiency is low and, as a result, laborious, tedious, timeconsuming multiple-step LLE procedures are usually needed to obtain acceptable recoveries. In this context, developing simple, fast, and efficient LLE strategies for FQ analysis has received extensive attention.
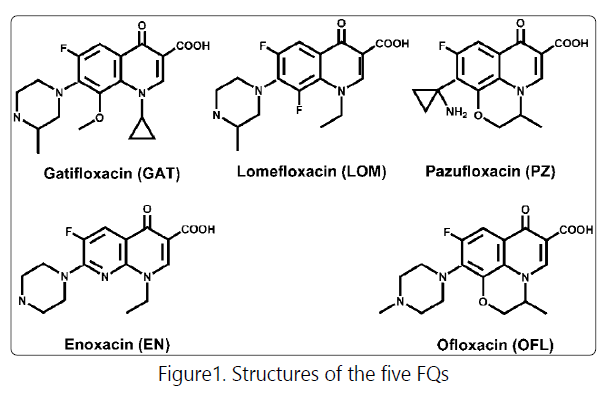
Ionic liquids (ILs) are a family of organic molten salts composed entirely of ions at room temperature. Their unique physical and chemical properties make them promising candidates for use as solvents, in many cases as extraction reagents in sample enrichment [14]. In this report, we synthesized an imidazolium-based IL 1-dodecyl-3-methylimidazolium bromide ([C12MIM]Br), using it as additive to the donor (aqueous) phase in LLE of five FQs, viz., gatifloxacin (GAT), lomefloxacin (LOM), enoxacin (EN), ofloxacin (OFL) and pazufloxacin (PZ) (structures are shown in Fig. 1), by dichloromethane. The parameters affecting the extraction recoveries were investigated and optimized. Finally, the IL-mediated LLE was coupled to CEUV for determination FQs in spiked milk samples.
Materials and Methods
Reagents
The five FQ antibiotics, namely, GAT, LOM, EN, OFL and
PZ, were purchased from Beijing Institute for Drug Control
(Beijing, China). The starting material 1-methylimidazole was
the product of Sigma (USA). Sodium hydroxide,
1-bromodecane, sodium dihydrogen phosphate, disodium
hydrogen phosphate, sodium tetraborate, dichloromethane
and n-butane were supplied by Beijing Chemical Reagents
(Beijing, China). Unless otherwise stated, deionized water
(Millipore, Milford, MA, USA) was used for preparing solutions.
The stock solutions of FQs at 1000 μg/mL each and ionic
liquids (20.0 g/L) were prepared in 6 mM sodium hydroxide
and deionized water, respectively. The stock solutions were stored in refrigerator at 4 °C and diluted properly before
experiments.
Preparation of [C12MIM]Br
[C12MIM]Br was synthesized according to the methods reported elsewhere [15-16]. Briefly, to a 250-mL round-bottomed flask, equal moles (0.2 mol) of 1-bromododecane and 1-methylimidazole were added, and the mixture was stirred at 80 °C for 24 hours to give a brown viscous liquid. After cooling, the product was washed three times with 50 mL of ethyl acetate. The washed product was dried at 70 °C to remove the organic solvent, with a yield of about 65%.
Capillary Electrophoresis
CE experiments were carried out on a CE system consisting
of an HB-Z303-1 AC high-voltage power supply (Hengbo,
Tianjin, China) and a CE-10 UV detector (Johnsson Separation
Science, Liaoning, China). The signal from the detector was
collected by a desktop PC via a CT22 data acquisition unit
(Qianpu, Jiangsu, China), then processed with HW2000
chromatography station (Qianpu). Separations were
conducted in a 60.0 cm (effective length = 50.0 cm) x 100 μm
i.d. fused-silica capillary (Yongnian Photoconduction Fibre,
Hebei, China). The running buffer comprised of 30 mM
sodium tetraborate and 10 mM sodium dihydrogen phosphate
at pH 8.5. The fresh capillary was treated sequentially with 0.2
M NaOH for 1 h, deionized water for 20 min, and the buffer
solution for 10 min. Samples were hydrostatically injected by
20 s at a height of 20.0 cm. The separation voltage was set at
10 kV, and the UV detection wavelength was set at 254 nm. To
determining the electroosmotic flow (EOF), dimethyl sulfoxide
(DMSO) was employed as the marker. The capillary was rinsed
with the running buffer for 2 min between two consecutive
runs. All the solutions used in CE were filtered using a 0.22 μm
filter (Jiuding, Beijing, China).
IL-Mediated LLE
To a 0.5 mL FQ standard solution, [C12MIM]Br was added
to a desired concentration (0, 0.5, 1.0, 2.0, 5.0 or 10.0 g/L). The
solution was stirred to mix thoroughly, and then stored in
darkness at 4 °C for 2 h. Afterward, the solution was mixed
with equal volume of dichloromethane, vortexed for 5 min,
followed by centrifugation at 4000 rpm for 5 min for phase
separation. The dichloromethane phase was placed in a 2 mL
centrifuge tube and was removed at reduced pressure using
a rotary evaporator (Seastar, Jiangsu, China). The FQ residues
were dissolved in 0.5 mL deionized water, and were subjected
to quantification by capillary electrophoresis-ultraviolet
detection (CE-UV). The recovery enhancement factor (REF),
defined as the ratio of recoveries with addition of ionic liquid
to that without addition of ionic liquid, was adopted to assess
the influence of ionic liquids on FQ extraction.
Determination of the Distribution Coefficient of IL in
Aqueous Phase
To evaluate the distribution of the IL [C12MIM]Br between
the aqueous phase (typically, 40 mM phosphate-tetraborate
at pH 8.2) and the dichloromethane phase, the parameter distribution coefficient was adopted, which was defined as
the ratio of IL concentration found in aqueous phase over the
initial concentration introduced. The IL cation ([C12MIM]+) in
aqueous phase was determined by capillary electrophoresis,
by using 50 mM phosphate at pH 6.5 as running buffer and by
monitoring [C12MIM]+ cation at 214 nm. For accurate
quantification, a calibration curve was established by CE
analysis of IL in the 40 mM phosphate-tetraborate solution at
varying concentration. The concentration of IL in aqueous
phase was calculated against the calibration curve.
Determination of Critical Micelle Concentration (CMC)
The CMC of [C12MIM]Br in 40 mM phosphate-tetraborate at pH 8.2 was determined by conductometric titration using a DDS-11A conductometer (Hongyi Instrumentation, Shanghai, China). Under vigorous stirring, 40 mL of 40 mM phosphatetetraborate at pH 8.2 was titrated with a solution containing 100 mM [C12MIM]Br and 40 mM phosphate-tetraborate at pH 8.2. The conductivity-IL concentration plots showed two straight lines with different slopes. The first one corresponded to the conductivity of monomers when the concentration was below CMC, while the second line with low slope at higher concentration corresponded to formation of micelles. The intersection was taken as the CMC value of [C12MIM]Br in this buffer solution.
Analysis of FQs in Fortified Milk Samples
The real sample of pure milk was purchased from a local supermarket. Four milliliters of milk was spiked with a mixture of FQs standards to desired concentrations (typically, 20 ng/ mL each), then was added [C12MIM]Br to 2.0 g/L and mixed thoroughly under vortex. After stored in refrigerator at 4 °C for 2 h, the sample was dilute to 10.0 mL with 40 mM phosphate buffer solution at pH 8.2. The mixture was agitated and then treated with ultrasonic (KQ-100, Jiangsu, China) for 5 min. Phase separation was obtained by centrifugation at 4000 r/min for 5 min. The supernatant was collected, and the solid residues were re-extracted with the same procedure. The two extracts were combined, added with 10.0 mL of dichloromethane, vortexed for 0.5 min and sonicated for 5 min. After the organic phase was collected, the aqueous phase was re-extracted with another 10 mL dichloromethane. The combined organic phases were evaporated at reduced pressure, and the FQs extracted were dissolved in 0.04 mL deionized water before analysis by CE.
Results and Discussion
Influence of Donor Phase Conditions
Effect of pH Value
Batch experiments were performed to investigate the
impact of aqueous phase pH on the recovery of extraction.
FQs were added to concentrations of 20 μg/L each in 40 mM
phosphate-tetraborate solutions containing 2.0 g/L [C12MIM]
Br at pH values of 5.80, 6.40, 7.20, 7.80, 8.20, 8.90, 9.68, and
10.78, respectively. The solutions were subjected to extraction
and CE analysis. The results (Fig. 2) showed that the peak
intensity of the extracted FQs increased with enhancing pH of the aqueous phase, reaching a maximum around pH 8.20, and
then decreased when the acidity of the donor phase was
further improved.
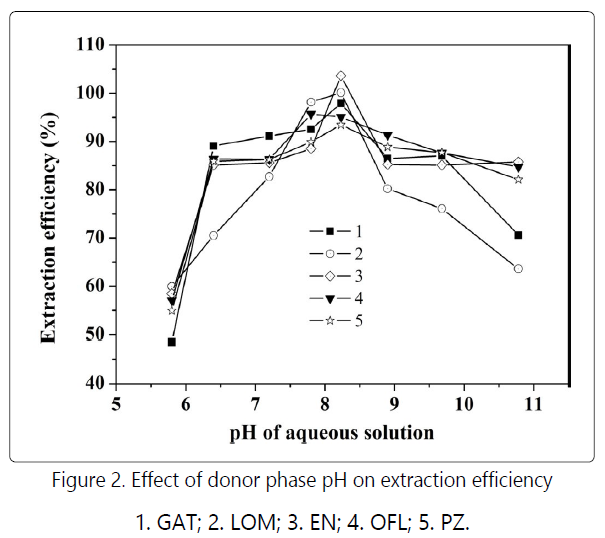
FQs are amphiphilic molecules containing both carboxyl and amino groups in their structures. The dissociation constant of carboxyl group (pKa1) is around 6, while the dissociation constant of the protonated amino groups (pKa2) is ca. 9. Therefore, the net charges of the FQ molecules were zero at pH 7-8 [17-18]. Under this circumstance, the FQs were dominantly hydrophobic, their distribution ratios in the organic phase being large. However, at higher or lower pH, the FQ molecules were charged, becoming hydrophilic and smaller distribution ratios in the organic phase would be observed. Not unexpectedly, It was found that under the same aqueous phase conditions, the extraction efficiency increased with addition of the ionic liquid [C12MIM]Br to the donor phase, which was predominately because the active C2-H on the imidazole ring tended to electrostatically interact and even from hydrogen bonds with the negatively charged groups of the other molecules 18. With regard to the FQs, the carboxyl and fluorine groups might be the active sites for the interactions. In addition, the hydrophobic long side chain on the imidazolium ring might also contribute the elevated recoveries because it could promote the distribution of FQs in the dichloromethane phase.
Impact of Ionic Strength
The influence of the aqueous phosphate-tetraborate
buffer concentration was investigated in the range of 10-60
mM at pH 8.2, with an interval of 10 mM. The recoveries of the
FQs increased with the buffer concentration till 40 mM,
beyond which the extraction efficiencies tended to drop. The
increase in the ionic strength could decrease the solubility of
the organic FQs (salting-out effect) and increase their
recoveries. On the other hand, high buffer concentration
would increase viscosity of the solution, resulting in low
molecular mass transfer rate and, consequently, low extraction
efficiency. In addition, high ionic strength also lowered the
CMC of the IL and therefore impacted negative influence on the extraction recovery. Based on the above experiment, the
donor phase was added with 40 mM phosphate-tetraborate
to adjust pH to 8.2.
Effect of IL Concentration
Fig. 3 depicts that REF initially increased with [C12MIM]Br
concentration, reaching the maximum at an IL concentration
of 2.0 g/L, whereby the REFs were between 1.3 and 2, or, in
the other words, the extraction efficiency increased by 30%
-100% as compared to the conventional dichloromethane
extraction method. However, as the IL concentration
continued to increase, REF gradually reduced. When the IL
concentration reaches 10.0 g/L, the REFs of the five FQs were
less than 1, indicating that the extraction efficiency under this
condition was lower than that of the conventional
dichloromethane extraction.
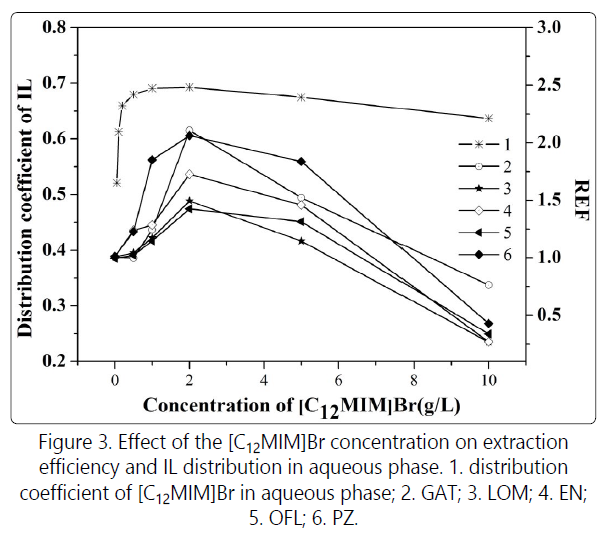
[C12MIM]Br is a cationic surfactant. At a concentration exceeding the critical micelle concentration (CMC), the imidazolium cations will form spherical micelles, with the hydrophilic imidazole groups pointing outward and the hydrophobic alkyl chains pointing to the core. The CMC of [C12MIM]+ in 20 mM phosphate solution at room temperature was reported to be 5 mM [19]. In our experiment, a 40 mM phosphate-tetraborate buffer solution was employed in which the CMC of [C12MIM]Br was determined to be 4.05 mM. To further explore the mechanism [C12MIM]Br affecting extraction efficiency, different quantities of the IL were introduced to the extraction system, and the distribution coefficients of [C12MIM]Br in the aqueous phase were determined (Fig. 3). The distribution coefficients of [C12MIM] Br in the aqueous phase increased with the IL concentration, from 0.521 at 0.05 g/L until 0.693 at 2 g/L, at which the [C12MIM]Br concentration in the aqueous phase was 1.38 g/L, equivalent to 4.16 mM. Although the distribution coefficients decreased slightly with the further increase in IL concentration (the values were 0.675 and 0.637 at 5 and 10 g/L, respectively), it was relatively stable.
At pH 8.2, FQs were partially negatively charged. After they electrostatically interact with the positively charged [C12MIM]+ (ion-pairing), the FQ-[C12MIM] complexes were nearly neutral, readily to distribute in organic phase, leading to enhanced extraction efficiencies. However, when the IL concentration was higher than its CMC in the aqueous phase, the FQs might first adsorb on the micelle surface as triggered by the electrostatic force, and then they might penetrate the outer hydrophilic micelle layer to the hydrophobic core, therefore, reducing the extraction efficiency. The dramatically lowered RTFs with the increasing IL concentration from 2 g/L implies that the hydrophobic interaction between the IL sidechain, and the FQ molecules was stronger than the ion-pairing effect, so, the influence of IL micelle-entrapping was strengthened with the improved concentration of micelles. Interestingly, Fig. 3 shows that in the presence of high concentration [C12MIM]Br, FQs might be stripped from the organic phase, indicating the wide potential application spectra of the ionic liquid [C12MIM]Br as an additive in LLE.
Performance of the Method
Capillary Electrophoresis
The assay linearity was evaluated by the analysis of five concentration levels of the FQ standard solutions from 5.0 to 100.0 μg/mL. Calibration curves, obtained by plotting peak areas (y, μV.s) versus the concentrations (μg/mL) of the standards, showed good linearity with correlation coefficient r2 ≥ 0.9990 (Table 1).
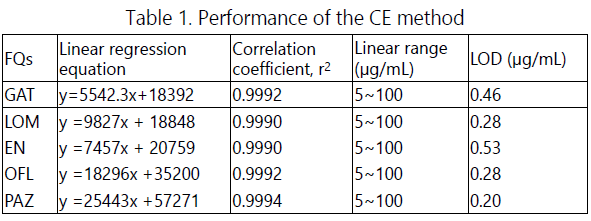
The limits of detection (LODs) determined as a S/N of 3, ranged from 0.20 to 0.53 μg/mL. The relative standard deviations (RSDs) of migration times and peak areas based on five consecutive runs were less than 3.07% and 8.03%, respectively.
Performance of IL-mediated LLE and Real Sample Analysis
Based on the optimal extraction conditions, i.e., the aqueous phase being composed of 40 mM phosphatetetraborate at pH 8.2, in the presence of 2 g/L [C12MIM]Br, and the acceptor phase being dichloromethane, recoveries of the FQs were assessed by spiking the FQs in a real milk sample to 0.10 μg/mL each. The IL-mediated LLE exhibited good recoveries, ranging from 74.45% to 106.75% (Table 2).
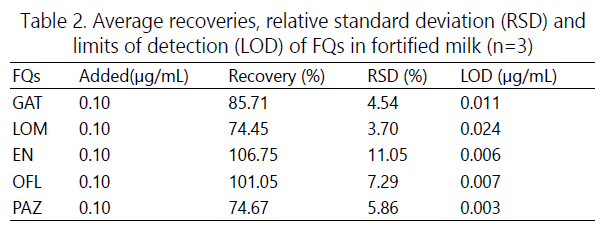
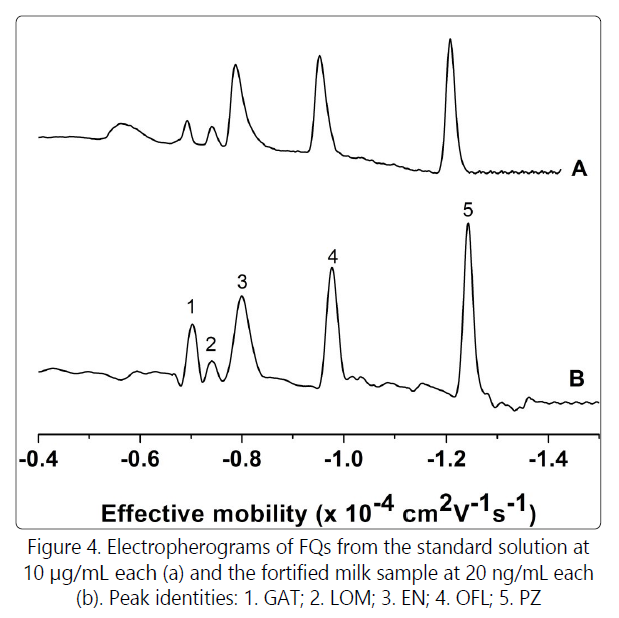
Due to the influence of the sample matrix, the intensity of the EOF varied significantly relative to that of standard solution. In order to improve the applicability of the method for real-world sample analysis, the electropherograms were reconstructed by plotting detector signal against effective mobilities (Fig. 4), from which one can observe that the effective mobilities of the FQs in real sample are consistent with those in the standard solution, and, consequently, the FQs could be easily identified. The effective mobility (μeff) was calculated by μeff =(1/teo-1/ti) xLl/V, where, ti is the migration time of analyte, and teo is the migration time of electroosmotic flow, L is the total length and l is the effective length of the capillary, and V is the applied voltage. Taking into consideration the extraction enrichment factor, the combinational strategy of IL-mediated LLE-CE-UV could offer LODs of 0.003 to 0.024 μg/mL (S/N=3) for FQs in milk samples. GAT, LOM, EN, OFL and PAZ are wildly prescribed antibiotics over the world. Based on the recommended MRL of 150 μg/kg for the FQs that are not listed in the EU Regulation [20], we suggest that the method developed has the potential for assaying FQs in samples with complex matrix. Importantly, the experimental results show that the addition of ionic liquid can significantly increase the single extraction efficiency. Compared with the report [21], the experimental procedure is greatly simplified, and the amount of organic solvent is substantially reduced.
Conclusions
In this work, an IL surfactant [C12MIM]Br was synthesized and investigated as an additive to the dichloromethanebased LLE of FQ antibiotics. Due to the hydrophobicity of the IL and due to the electrostatic interaction and hydrogen bonding between the IL and the FQ molecules, addition of IL to the donor phase gave rise to improved recoveries of the FQs. The IL-mediated LLE method had a number of advantages, including less usage of organic solvents, speediness, high extraction efficiency and ease to operate. The CE-UV method could baseline separate the FQs with high sensitivity. Moreover, reconstructing the electropherogram by plotting the detector signal versus effective mobility could eliminate the influence of background matrices on the identification, improving the applicability of the method for real sample analysis. Our results suggested that hyphening IL-mediated LLE to CE-UV can offer quick and accurate determination of trace FQs in complex matrices.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21575017) and the Fundamental Research Funds for the Central Universities.
Conflicts of Interest
The authors have declared no conflict of interest.
References
- Janssen H, Janssen I, Cooper P, et al. Antimicrobial-Resistant Bacteria in Infected Wounds, Ghana, 2014. Emerg. Infect. Dis. 2018; 24(5): 916-919. doi: 10.3201/eid2405.171506
- Commission regulation (EU) No. 37/2010 of 22 December 2009 on pharmacologically active substances and their classification regarding maximum residue limits in foodstuffs of animal origin.
- Announcement No. 235 of Ministry of Agriculture, The national standards for maximum residue level of veterinary drugs in food of animal origin (2002), Ministry of Agriculture of People's Republic of China.
- da Silva WP, de Oliveira LH, dos Santos AL, Ferreira VS, Goncalves Trindade MA. Sample preparation combined with electroanalysis to improve simultaneous determination of antibiotics in animal derived food samples. Food Chem. 2018; 250: 7-13. doi: 10.1016/j.foodchem.2018.01.041
- Takeda N, Gotoh M, Matsuoka T. Rapid screening method for quinolone residues in livestock and fishery products using immobilised metal chelate affinity chromatographic clean-up and liquid chromatographyfluorescence detection. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. 2011; 28(9): 1168-1174. doi: 10.1080/19440049.2011.587028
- Zhu M, Zhao H, Xia D, Du J, Xie H, Chen J. Determination of 21 antibiotics in sea cucumber using accelerated solvent extraction with in-cell clean-up coupled to ultra-performance liquid chromatography-tandem mass spectrometry. Food Chem. 2018; 258: 87-94. doi: 10.1016/j. foodchem.2018.03.051
- Chiesa L, Panseri S, Pasquale E, Malandra R, Pavlovic R, Arioli F. Validated multiclass targeted determination of antibiotics in fish with high performance liquid chromatography-benchtop quadrupole orbitrap hybrid mass spectrometry. Food Chem. 2018; 258: 222-230. doi: 10.1016/j. foodchem.2018.03.072
- Meng R, Kang J. Determination of the stereoisomeric impurities of sitafloxacin by capillary electrophoresis with dual chiral additives. J. Chromatogr. A. 2017; 1506: 120-127. doi: 10.1016/j.chroma.2017.05.010
- Zhang X-H, Deng Y, Zhao M-Z, Zhou Y-L, Zhang X-X. Highly-sensitive detection of eight typical fluoroquinolone antibiotics by capillary electrophoresis-mass spectroscopy coupled with immunoaffinity extraction. Rsc Advances. 2018; 8(8): 4063-4071. doi: 10.1039/C7RA12557G
- Turiel E, Bordin G, Rodriguez AR. Trace enrichment of (fluoro)quinolone antibiotics in surface waters by solid-phase extraction and their determination by liquid chromatography-ultraviolet detection. J. Chromatogr. A. 2003; 1008(2): 145-155. doi: 10.1016/S0021-9673(03)00975-0
- Monteiro MA, Spisso BF, Ferreira RG, et al. Development and Validation of Liquid Chromatography-Tandem Mass Spectrometry Methods for Determination of Beta-Lactams, Macrolides, Fluoroquinolones, Sulfonamides and Tetracyclines in Surface and Drinking Water from Rio de Janeiro, Brazil. Journal of the Brazilian Chemical Society. 2018; 29(4): 801-813. doi: 10.21577/0103-5053.20170203
- Vakh C, Alaboud M, Lebedinets S, et al. An automated magnetic dispersive micro-solid phase extraction in a fluidized reactor for the determination of fluoroquinolones in baby food samples. Anal. Chim. Acta. 2018; 1001: 59-69. doi: 10.1016/j.aca.2017.11.065
- Timofeeva I, Timofeev S, Moskvin L, Bulatov A. A dispersive liquid-liquid microextraction using a switchable polarity dispersive solvent. Automated HPLC-FLD determination of ofloxacin in chicken meat. Anal. Chim. Acta. 2017; 949: 35-42. doi: 10.1016/j.aca.2016.11.018
- Reyna-Gonzalez JM, Torriero AAJ, Siriwardana AI, Burgar IM, Bond AM. Extraction of Copper(II) Ions from Aqueous Solutions with a Methimazole- Based Ionic Liquid. Anal. Chem. 2010; 82(18): 7691-7698. doi: 10.1021/ Ac101321a
- Latep N, Liang X, Qin W. Ionic-liquid-assisted desorption of DNA from polyamidoamine-grafted silica nanoparticles surface by a low-salt solution. J. Sep. Sci. 2014; 37(15): 2069-2076. doi: 10.1002/jssc.201400203
- Tursen J, Wang A, Qin W. Electrophoretic separation of acidic and basic proteins in the presence of micromolar concentrations of an ionic liquid. Microchim. Acta. 2011; 174(1-2): 63-71. doi: 10.1007/s00604-011-0599-y
- See KL, Elbashir AA, Saad B, Ali ASM, Aboul-Enein HY. Simultaneous determination of ofloxacin and ornidazole in pharmaceutical preparations by capillary zone electrophoresis. Biomed. Chromatogr. 2009; 23(12): 1283-1290. doi: 10.1002/bmc.1251
- Chen Z, Zhong Z, Xia Z, Yang F, Mu X. Separation of Fluoroquinolones by MEKC Modified with Hydrophobic Ionic Liquid as a Modifier. Chromatographia. 2011; 75(1-2): 65-70. doi: 10.1007/s10337-011-2150-z
- Borissova M, Palk K, Koel M. Micellar electrophoresis using ionic liquids. J. Chromatogr. A. 2008; 1183(1-2): 192-195. doi: 10.1016/j.chroma.2007.12.077
- Toussaint B, Chedin M, Bordin G, Rodriguez AR. Determination of (fluoro) quinolone antibiotic residues in pig kidney using liquid chromatographytandem mass spectrometry I. Laboratory-validated method. J. Chromatogr. A. 2005; 1088(1-2): 32-39. doi: 10.1016/j.chroma.2005.02.057
- Fan Y, Gan X, Li S, Qin W. A rapid CE-potential gradient detection method for determination of quinolones. Electrophoresis. 2007; 28(22): 4101-4107. doi: 10.1002/elps.200700182
