

Research Article
Silica nanoparticle-mediated, Electric fieldtriggered sensitivity enhancement strategy for Capillary electrophoretic detection of proteins
Beijing Normal University, Beijing, China
*Corresponding author: Weidong Qin, Key Laboratory of Theoretical and Computational Photochemistry, Ministry of Education, College of Chemistry, Beijing Normal University, Beijing 100875, China Fax: + 86-10-58802531 Email: qinwd@bnu.edu.cn
Received: October 27, 2016 Accepted: November 10, 2016 Published: November 14, 2016
Citation: Wang A, Qin W. Silica nanoparticlemediated, Electric field-triggered sensitivity enhancement strategy for Capillary electrophoretic detection of proteins. Madridge J Anal Sci Instrum. 2016; 1(1): 11-15. doi: 10.18689/mjai-1000103
Copyright: © 2016 The Author(s). This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
We report in this paper the improved capillary electrophoresis (CE)-UV detection sensitivity of proteins based on silica nanoparticle-mediated, electric field-triggered (SNM-EFT) strategy. During the process, the capillary was first filled with acidic separation buffer, followed by injection of a segment of alkaline treating solution containing silica nanoparticles (SNPs). After the inlet end of the capillary was dipped into the protein solution containing SNPs, a high voltage was applied across the capillary. The SNPs in the protein solution became aggregate, promoting the SNP-protein conjugation, by which transformation of the protein structures took place. During CE, the unfolded protein desorbed from SNPs, generating stronger signal than the native one due to the more effective exposure of tryptophanyl residues to the polar buffer. The parameters of the treatment protocols, e.g., pH of the treating solution, concentration of the SNPs presented, the voltage and duration of the electric field applied, had notable effects on the detection sensitivity. Significant improvement in CE-UV response was obtained by the EFT at -8 kV × 60 s on the protein standards dissolved in 4 mM sodium tetraborate, 1 mM boric acid and 0.015% SNPs.
Keywords: Capillary electrophoresis; Proteins; Silica nanoparticles; Electric field; sensitivity
Abbreviations: SNPs, silica nanoparticles; LZ, lysozyme; BSA, bovine serum albumin; HB, hemoglobin; PEO, polyethylene oxide; CE, capillary electrophoresis
Introduction
Nanoparticles (NPs) can conjugate with proteins via hydrophobic patches, hydrogen
bonding and columbic interactions owing to the large surface area of NPs [1,2] and to
the simultaneous presence of hydrophobic, hydrophilic, cationic, and anionic groups at
the surface of protein molecules [3]. The NP-protein conjugation is size-dependent,
higher degree conjugation takes place with aggregated NPs [4]. Moreover, such
interaction often results in tertiary conformational transformation (unfolding) of the
protein [5-8].
Capillary electrophoresis (CE) combines well-known advantages of speedy
separation, high efficiency, small amount of sample and solvent consumption, and high
automation [9-12]. Due to these unique advantages, it has been extensively employed
in proteome research. Not surprisingly, NPs have been hyphenated to CE [13-17] and
chip-based electrophoresis system [18-20] for protein separation. For example, a buffer
containing surfactant-capped gold nanoparticles (AuNPs) allowed simultaneous
separation of acidic and basic proteins in a single run [13,14]. By employing lipid-based liquid crystalline NPs as pseudostationary phase (PSP), green
fluorescent protein (GFP) and its mutants were baseline
separated at neutral pH [17]. Moreover, silica nanoparticles
(SNPs)acting as PSPs could also render enhanced resolution
for proteins [16]. Liu and co-workers reported TiO2 NP-coated
open-tubular capillary electrochromatographic separation of
proteins [15], in which conalbumin (ConA) and apo-transferrin
(apoTf) of similar molecular weight could be baseline
separated.
However, in contrast to the large number of report
enhancing the resolution, few reports were dedicated to
investigate their applicability in sensitive detection of proteins
by CE. The main reason might be that both the fluorescence
emission and UV absorbance decrease upon adsorption of
protein to nanoparticles [7,21]. Nonetheless, such NP-protein
conjugation might be adopted under some circumstances for
sensitive protein detection. Studies revealed that the refolding
rates of some proteins, such as lysozyme [22], are very slow
after deconjugation. The surrounding environments of the
amino acid residues in the detached, unfolding proteins might
be different than those in the normal proteins. In this context,
the UV-active amino acid residues, i.e., tyrosine, tryptophan
and phenylalanine, might have different molar absorptivities
which would lead to different detection sensitivities. This
strategy can be readily realized in CE techniques due to its
quick analysis and ease in tuning the chemical properties of
the running buffer to facilitate the deconjugation under the
electric field.
The aim of this work is to demonstrate the proof-ofprinciple
application of the silica nanoparticle-mediated,
electric field-triggered (SNM-EFT) sensitivity enhancement
strategy for CE-UV detection of unfolded proteins following
the decomposition of the SNP-protein complexes. SNPs were
employed as model nanoparticles for investigation. To
initialize the SNP-protein complexation, a capillary was first
filled with separation buffer followed by injection of a segment
of alkaline treating solution containing SNPs. After the inlet
end of the capillary was dipped into the protein solution
containing SNPs, a high voltage was applied across the
capillary. A low conductivity zone formed at the interface
between the acidic CE buffer and the alkaline treating solution
due to neutralization, whereby the SNPs aggregated, moving
to the injection vial and promoting the SNP-protein
conjugation. The SNM-EFT parameters, e.g., pH and the SNPs
concentration of the treating solution, the electric voltage
and duration of EFT, were investigated. Notable improvement
in sensitivity was observed for UV detection of the model
proteins, especially for lysozyme, suggesting the potential of
the strategy in CE analysis of proteins.
Materials and methods
Reagents and solutions
Reagents
Hemoglobin (HB) and lysozyme (LZ) were from Sigma (St.
Louis, MO, USA); bovine serum albumin (BSA, section V) was
purchased from Amresco (Solon, OH, USA). PEO (polyethylene oxide, Mr 1 000 000) was supplied by Alfa Aesar (Ward Hill,
MA, USA). The SNPs with average diameter of 20 nm and
purity of 99.9% were bought from Nanjing Nano High-Tech
(Jiangsu, China). All the other chemicals were of analytical
grade. Double-distilled water was used to prepare buffers and
solutions throughout the experiment.
Solutions
SNP suspension at 1% (w/v) was prepared by gradually
adding precisely weighed SNPs to double-distilled water
under vigorous stirring. Stock solutions of 100 mM phosphoric
acid, 100 mM sodium tetraborate and 100 mM boric acid were
employed for preparing buffer solutions of desired
concentrations and pH values. Working solutions of protein
standards (mixture of 50 mg•L-1 each) were prepared by
mixing the individual stock solutions (10000 mg•L-1 in doubledistilled
water) with appropriate volumes of borate buffer,
SNPs suspension and diluting with double-distilled water.
Capillary electrophoresis
The CE-UV system consisted of a DW-P303-1AC capillary
electrophoresis high-voltage power supply (Sanchuan High
Tech, China) and a CE-10UV detector (Johnsson Separation
Science, Liaoning, China) operated at 210 nm. Signal from the
detector was acquired and processed with HW2000
chromatography station (Qianpu, Jiangsu, China). A 50-cm
long polyimide-coated fused-silica capillary (40 cm in effective
length) of 75-µm i.d. and 375-µm o.d. (Yongnian
Photoconduction Fibre, Hebei, China) was used for CE. The
fresh capillary was consecutively rinsed for 30 min with 1 M
NaOH, 10 min with double-distilled water and 5 min with
separation buffer. Each electrolyte solution was filtered
through a 0.22-µm membrane filter (Jiuding High Tech,
Beijing, China). Samples were hydrodynamically injected into
the capillary by raising the inlet reservoir 20.0 cm for 30 s.
Electrophoretic separations were carried out at a voltage of
11 kV under ambient temperature.
SNM-EFT procedure
The capillary was first rinsed for 2 min with the separation
buffer, followed by hydrodynamic injection of a segment of
treating solution by siphon (the optimum parameters were 20
cm ´ 40 s). After that, the inlet was immersed into the sample
vial containing the protein standard solution and the outlet
was immersed into the separation buffer. A negative voltage
was then applied between the sample and the outlet vials for
a desired duration. The capillary was flushed with running
buffer for 2 min after the procedure, and the treated protein
solution was vigorously shaken before injection.
Results and discussion
Optimization of CE conditions
Background electrolyte (BGE) of 20 mM phosphoric acid
(pH 1.89) was employed to suppress the deprotonation of
silanol groups of the capillary inner wall and, therefore, to
suppress the wall-adsorption of the positively charged
proteins. In order to further improve the separation performances, PEO was added to the buffer, and its
concentration was optimized. Improved peak intensities were
observed in the presence of 0.05% SNPs (Figure 1B vs. 1A).
The buffer containing 0.2% PEO offered the highest resolutions
for the proteins (Figure 1C) although the overall sensitivity of
the three proteins was slightly lower than that of Figure 1B.
Addition of PEO to the buffer increases the buffer viscosity,
resulting in reduced injection volume and consequently the
decreased peak heights of the analytes. Taking into account
the parameters of resolution and sensitivity, we chose BGE
consisting 20 mM phosphoric acid and 0.2% PEO for the
further experiments.
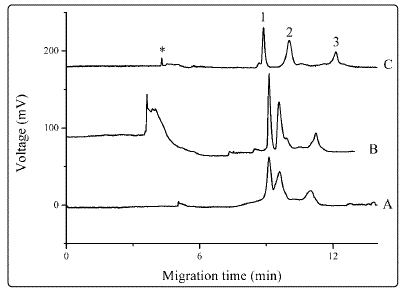
Figure 1. Influence of buffer additives Separations were carried out with BGE of 20 mM phosphoric acid (A), which was added to 0.05% SNPs (B) or 0.2% PEO (C). Proteins were dissolved to concentrations of 50 mg•L-1 each in treating solution. Electrophoresis was conducted at 11 kV and UV detection was performed at 210 nm. Peak identities: 1, LZ; 2, BSA; 3, HB; *, systemic peak. Electropherograms were offset for clarity.
Preliminary experiments on SNM-EFT
Experiments were carried out to explore the influence of
SNPs and electric field on the detection sensitivity. Two kinds
of treating solutions were employed, i.e., 4 mM sodium
tetraborate and 1 mM boric acid at pH= 9.11 with and without
0.015% SNPs, they are denoted treating solutions A and B,
respectively. Our preliminary experiments suggested that
application of positive high voltage did not change the
detection sensitivity of the proteins (Figure 1S of the
Supporting Information); so, a negative voltage was employed.
We initially employed same treating solutions for injecting
into the capillary and for dissolving the protein standards.
Compared to treating solution A (Figure 2A, without EFT),
using treating solution B did not result in higher solution; on
the contrary, lower peak heights (Fig, 2B, without EFT) were
observed, probably due to the higher solution viscosity in the
presence of 0.015% SNPs. With EFT, utilizing treating solution
A (Figure 2C) did not bring about noticeable changes in
detection sensitivity as compared with Figure 2A. However,
when EFT was applied to treating solution B, remarkable
improvement of the peak heights was obtained, especially for
lysozyme. The results suggest the potential of SNM-EFT
strategy in sensitive detection of proteins.
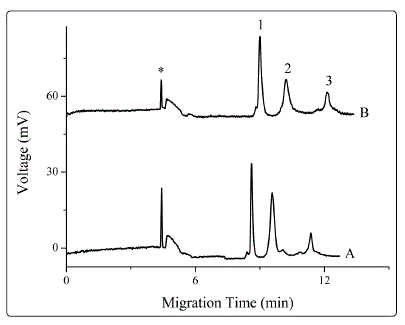
Figure 1S. Effect of positive high voltage on detection sensitivity. Sample treatment techniques: Proteins dissolved in 4 mM sodium tetraborate, 1 mM boric acid and 0.015% SNPs; the solution was treated for 60s at a voltage of (A) 0 kV; (B) +8 kV.
For better understanding the mechanisms, the SNM-EFT experiments were conducted using different treating solutions for pre-injection and for dissolving protein standards. With pre-injection of treating solution A, the detection sensitivities of the proteins (Figure 2E) were similar to those in Figure 2B. Likewise, peak heights of the proteins close to Figure 2A were observed with pre-injection of treating solution B.
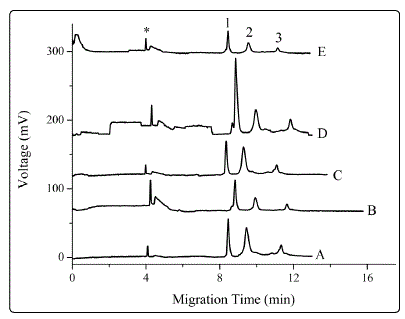
Figure 2. Effect of sample treatment techniques on detection sensitivity. Separation buffer: 20 mM phosphoric acid and 0.2% PEO. Peak identities: 1, LZ; 2, BSA; 3, HB; *, systemic peak. Preparation and treatment of the standards: (A) Proteins dissolved in treating solution A, without EFT; (B) proteins dissolved in treating solution B, without EFT; (C) proteins dissolved in treating solution A; EFT: -8 kV × 60s; (D) proteins dissolved in treating solution B, EFT: -8 kV × 60s. In (A)-(D), the plug of treating solution in each trace, introduced by 20 cm ´ 40s, was the same with that used in preparing the corresponding protein standards. (E) proteins were dissolved in treating solution B; treating solution A was injected into the capillary (20 cm ´ 40s); EFT: 8 kV × 60s. Other conditions were the same as those in Figure 1.
During EFT process, a pH junction was formed at the boundary between the alkaline treating solution and the acidic separation buffer. The hydrogen ion (H+) from the BGE intruded into the treating solution plug under electric field and reacted with OH- and B(OH)4-, forming a low-conductivity zone where SNPs began to aggregate [23,24]. These aggregates were carried into the cathodic sample vial by EOF, broadening the primary particle size distribution (PPSD) and consequently accelerating the aggregating rate of the SNPs [25], promoting the SNP-protein conjugation.
Effect of SNP concentration and pH
Figure 3 depicts that under an EFT treatment of -8 kV ×
60 s, the peak height of LZ increases gradually with the
increasing concentration of SNPs until 0.015%, from which the
peak height kept at a relatively stable level. However, the peak
height of HB did not show significant improvement in the
presence of SNPs; moreover, addition of SNPs even caused
slightly decreased response of BSA. We suggest that presence
of SNPs improves the viscosity of the sample solution, resulting
in low injection volume and, hence, low detector response.
Therefore, the concentration of SNPs was kept at 0.015%.
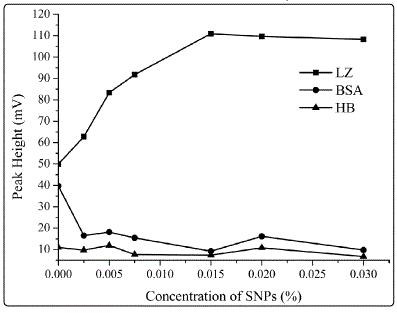
Figure 3. Dependence of peak heights on the concentration of SNPs presented in the treating solution. The protein concentrations were 50 mg•L-1 each. The treating solution in the experiment contained 4 mM sodium tetraborate, 1 mM boric acid and varying concentrations of SNPs from 0 to 0.03%. The other conditions were the same as those in Figure 2
To study the influence of treating solution pH, proteins
were dissolved in different treating solutions of 4 mM H3PO4
(pH= 2.11), 10 mM NaH2PO4 (pH= 4.27) and 4 mM sodium
tetraborate (pH= 9.11). All solutions were added with SNPs to
0.015%. Application of electric field did not lead to enhanced
detection sensitivity with proteins dissolved in acidic treating
solution; but it did in alkaline solutions.
Adsorption of LZ onto SNPs was influenced by the
nanoparticle size and solution pH. Multilayer adsorption and
greater conformational change occurred with proteins
attached on SNPs of larger size [4]. High solution pH promotes
these progresses [4].
Effect of electric voltage and treating duration
Significant enhancement in the response of LZ was
observed at negative voltages ranged from -8 to -10 kV (Figure
4). LZ is positively charged in buffer of pH 9.11 because its isoelectric point is ca 11. The results reveal that the
complementary electrostatic interaction facilitates the
adsorption of LZ to SNPs [4,22]. It is interesting to find that the
negatively charged BSA and HB, which should be electrostatically
repulsive to the SNPs, also indicate considerable enhancement
in detection sensitivity. The hydrophobic interaction might be
responsible for the adsorption of proteins to SNPs under this
circumstance [7]. Moreover, it was reported [26] that BSA and
HB possess low internal stability; they are “soft” and are prone
to adsorb on all surfaces irrespective of electrostatic interactions.
Figure 4. Effect of electric voltage on detection sensitivity. The sample solutions underwent EFT treatments for 60s at the following voltages: (A) 0 kV; (B) -5 kV; (C) -7 kV; (D) -8 kV; (E) -9 kV; (F) -10 kV; (G) -11 kV; (H) -12 kV. Figures in the inset: dependence of peak heights of proteins on the electric voltages. The other conditions were the same as those in Figure 2.
The treating time is another important factor influencing
the sensitivity; the peak height of LZ increased with the
duration first to a maximum at 60 s, then decreased with
further extended duration.
In the SNM-EFT strategy, high voltage generates high
EOF intensity and large amount of SNP aggregates, favoring
high sensitivity. Nevertheless, high migration velocities of the
ions in the capillary disturb the pH junction and, even worse,
under some circumstances, for example, the long EFT time,
the acidic buffer may enter the sample vial. The former does
not favor formation of aggregated SNPs, whereas the latter
will weaken the protein-SNP interaction [4,27].
Conclusions
We report the proof-of-principle application of SNM-EFT strategy for sensitive CE-UV detection of proteins. Influences of SNP concentration and pH of the treating solution, the treating electric voltage and duration were investigated. The SNP aggregates generated during the EFT promoted the aggregation of the SNPs in the standard solution and, as a result, favored the SNP-protein interaction. The unfolded protein desorbed from the SNPs during CE could produce enhanced UV-absorbance signal. Due to the great variety of nanoparticles and the wide pH range of the CE buffers employed in proteome research, we expect the method opens up new opportunities for sensitive detection of more proteins.
Acknowledgements
This work was supported by the National Natural Science
Foundation of China (21575017) and the Fundamental
Research Funds for the Central Universities.
Conflicts of Interest: The authors have declared no
conflict of interest.
References
- Assfalga M, Ragonab L, Paganob K, D′Onofrioa M, Zanzonia S, Tomasellib S, et al. The study of transient protein-nanoparticle interactions by solution NMR spectroscopy. Bba-Proteins Proteom. 2016;1864(1):102- 14. https://dx.doi.org/10.1016/j.bbapap.2015.04.024
- Kelly PM, Aberg C, Polo E, O′Connell A, Cookman J, Fallon J, et al. Mapping protein binding sites on the biomolecular corona of nanoparticles. Nature Nanotechnology. 2015;10(5):472-479. doi: 10.1038/nnano.2015.47
- Manabe T. Capillary electrophoresis of proteins for proteomic studies. Electrophoresis. 1999;20(15-16):3116-21. doi: 10.1002/(SICI)1522- 2683(19991001)20:15/16<3116::AID-ELPS3116>3.0.CO;2-0
- Vertegel AA, Siegel RW, Dordick JS. Silica nanoparticle size influences the structure and enzymatic activity of adsorbed lysozyme. Langmuir. 2004;20(16):6800-6807. doi: 10.1021/la0497200
- Czeslik C, Winter R. Effect of temperature on the conformation of lysozyme adsorbed to silica particles. Phys Chem Chem Phys. 2001;3(2):235-9. doi: 10.1039/B005900P
- Brewer SH, Glomm WR, Johnson MC, Knag MK, Franzen S. Probing BSA binding to citrate-coated gold nanoparticles and surfaces. Langmuir. 2005;21(20):9303-7. doi: 10.1021/la050588t
- Shang L, Wang YZ, Jiang JG, Dong SJ. pH-dependent protein conformational changes in albumin : gold nanoparticle bioconjugates: A spectroscopic study. Langmuir. 2007;23(5):2714-21. doi: 10.1021/ la062064e
- You CC, De M, Han G, Rotello VM. Tunable inhibition and denaturation of alpha-chymotrypsin with amino acid-functionalized gold nanoparticles. J Am Chem Soc. 2005;127(37):12873-81. doi: 10.1021/ ja0512881
- Heemskerk AAM, Deelder AM, Mayboroda OA. CE-ESI-MS for bottomup proteomics: Advances in separation, interfacing and applications. Mass Spectrom Rev. 2016;35(2):259-71. doi: 10.1002/mas.21432
- Chen J, Ni Y, Liu CC, Yamaguchi Y, Chen Q, Sekine S et al. Rapid identification and quantitation for oral bacteria based on short-end capillary electrophoresis. Talanta. 2016;160:425-30. doi: 10.1016/j. talanta.2016.07.049
- Mendes TD, Porto BLS, Bell MJV, Perrone ÍT, de Oliveira MA. Capillary zone electrophoresis for fatty acids with chemometrics for the determination of milk adulteration by whey addition. Food Chem. 2016;213:647-53. doi: 10.1016/j.foodchem.2016.07.035
- Riley KR, Liu S, Yu G, Libby K, Cubicciotti R, Colyer CL. Using capillary electrophoresis to characterize polymeric particles. J Chromatogr A. 2016;1463:169-75. doi: 10.1016/j.chroma.2016.08.017
- Yu CJ, Su CL, Tseng WL. Separation of acidic and basic proteins by nanoparticle-filled capillary electrophoresis. Anal Chem. 2006;78(23):8004-10. doi: 10.1021/ac061059c
- Lin CY, Liu CH, Chang HC, Tseng WL. Enrichment and separation of acidic and basic proteins using the centrifugal ultrafiltration followed by nanoparticle-filled capillary electrophoresis. Electrophoresis. 2008;29(14):3024-31. doi: 10.1002/elps.200700879
- Hsieh YL, Chen TH, Liu CY. Capillary electrochromatographic separation of proteins on a column coated with titanium dioxide nanoparticles. Electrophoresis. 2006;27(21):4288-94. doi: 10.1002/elps.200500897
- Qin WD. Silica nanoparticles as pseudostationary phase for protein separation. Electrophoresis. 2007;28(17):3017-23. doi: 10.1002/ elps.200600476
- Nilsson C, Becker K, Harwigsson I, Bülow L, Birnbaum S, Nilsson S. Hydrophobic Interaction Capillary Electrochromatography of Protein Mutants. Use of Lipid-Based Liquid Crystalline Nanoparticles as Pseudostationary Phase. Anal Chem. 2009;81(1):315-21. doi: 10.1021/ ac8020533
- Dou YH, Bao N, Xu JJ, Meng F, Chen HY. Separation of proteins on surface-modified poly(dimethylsiloxane) microfluidic devices. Electrophoresis. 2004;25(17):3024-31. doi: 10.1002/elps.200405986
- Wang AJ, Xu JJ, Chen HY. Proteins modification of poly(dimethylsiloxane) microfluidic channels for the enhanced microchip electrophoresis. J Chromatogr A. 2006;1107(1-2):257-64. https://dx.doi.org/10.1016/j. chroma.2005.12.040
- Kim HR, Andrieux K, Delomenie C, et al. Analysis of plasma protein adsorption onto PEGylated nanoparticles by complementary methods: 2-DE, CE and protein Lab-on-chip((R)) system. Electrophoresis. 2007;28(13):2252-61.
- Sun CX, Wu X, Ding HH, Zhao L, Wang F, Yang J. The Fluorescence Enhancement of the Protein Adsorbed on the Surface of Ag Nanoparticle. J Fluoresc. 2009;19(1):111-7. doi: 10.1007/s10895-008-0392-4
- Wu XY, Narsimhan G. Effect of surface concentration on secondary and tertiary conformational changes of lysozyme adsorbed on silica nanoparticles. Bba-Proteins Proteom. 2008;1784(11):1694-701. doi: 10.1016/j.bbapap.2008.06.008
- Prasher R, Phelan PE, Bhattacharya P. Effect of aggregation kinetics on the thermal conductivity of nanoscale colloidal solutions (nanofluid). Nano Lett. 2006;6(7):1529-34. doi: 10.1021/nl060992s
- Fedeyko JM, Vlachos DG, Lobo RF. Formation and structure of selfassembled silica nanoparticles in basic solutions of organic and inorganic cations. Langmuir. 2005;21(11):5197-206. doi: 10.1021/la0468390
- Peng ZB, Doroodchi E, Evans GM. Influence of primary particle size distribution on nanoparticles aggregation and suspension yield stress: A theoretical study. Powder Technol. 2012;223:3-11. https://dx.doi. org/10.1016/j.powtec.2011.11.001
- Lucy CA, MacDonald AM, Gulcev MD. Non-covalent capillary coatings for protein separations in capillary electrophoresis. J Chromatogr A. 2008;1184(1-2):81-105. https://dx.doi.org/10.1016/j.chroma.2007.10.114
- Haynes CA, Sliwinsky E, Norde W. Structural and electrostatic properties of globular proteins at a polystyrene-water interface. J Colloid Interface Sci. 1994;164(2):394-9. doi: 10.1006/jcis.1994.1182
